
Rapid adoption of molecular testing in cancer necessitates quality cell line-derived controls.
By Keith Cannon and Prabha Nagarajan, PhD
Molecular diagnostics can be broadly defined as the detection of biomarkers in an individual to study human diseases like cancer, infectious diseases, congenital abnormalities, etc. Here, we will focus on cancer diagnostics using genetic biomarkers; however, the underlying principles of genetic testing and sources of error are applicable to other diagnostic molecular assays, too. With the rapid growth and adoption of molecular testing in cancer, there are some major limitations related to the quality of analyte, the accuracy and sensitivity of analytical methods, and the reliability and reproducibility of the results. This calls for development and use of quality controls to calibrate measurements and establish sensitivities of the given analytical assays.
Genetic studies for cancer patients can be classified into cytogenetics and molecular genetics. Major chromosomal aberrations can be detected by cytogenetics; however, a deeper insight into specific mutations at the molecular level is important for differential diagnosis, prognosis, and disease management.
A tumor mutational profile is comprised mainly of single nucleotide variants (SNVs), which lead to amino acid changes, splicing alterations, or premature truncation of proteins. Small insertions or deletions (indels), gene copy number variations, and structural anomalies like translocations and fusions are other common types of DNA aberrations found in the tumor genome.
Tumor profiling tests can involve either a single gene/variant detection or a more complex test that involves detection of multiple SNVs and other types of mutations. Comprehensive genomic profiling involving simultaneous testing of multiple genes is increasingly popular. Multiple gene analysis gives better diagnostic yield and helps stratify patients into different risk groups, allowing targeted treatment. Next-generation sequencing (NGS) is widely used in oncology for its utility in investigating the underlying molecular mechanisms that cause cancer and for detection of variants in multiple genes simultaneously from patient samples.
Challenges in NGS Workflows
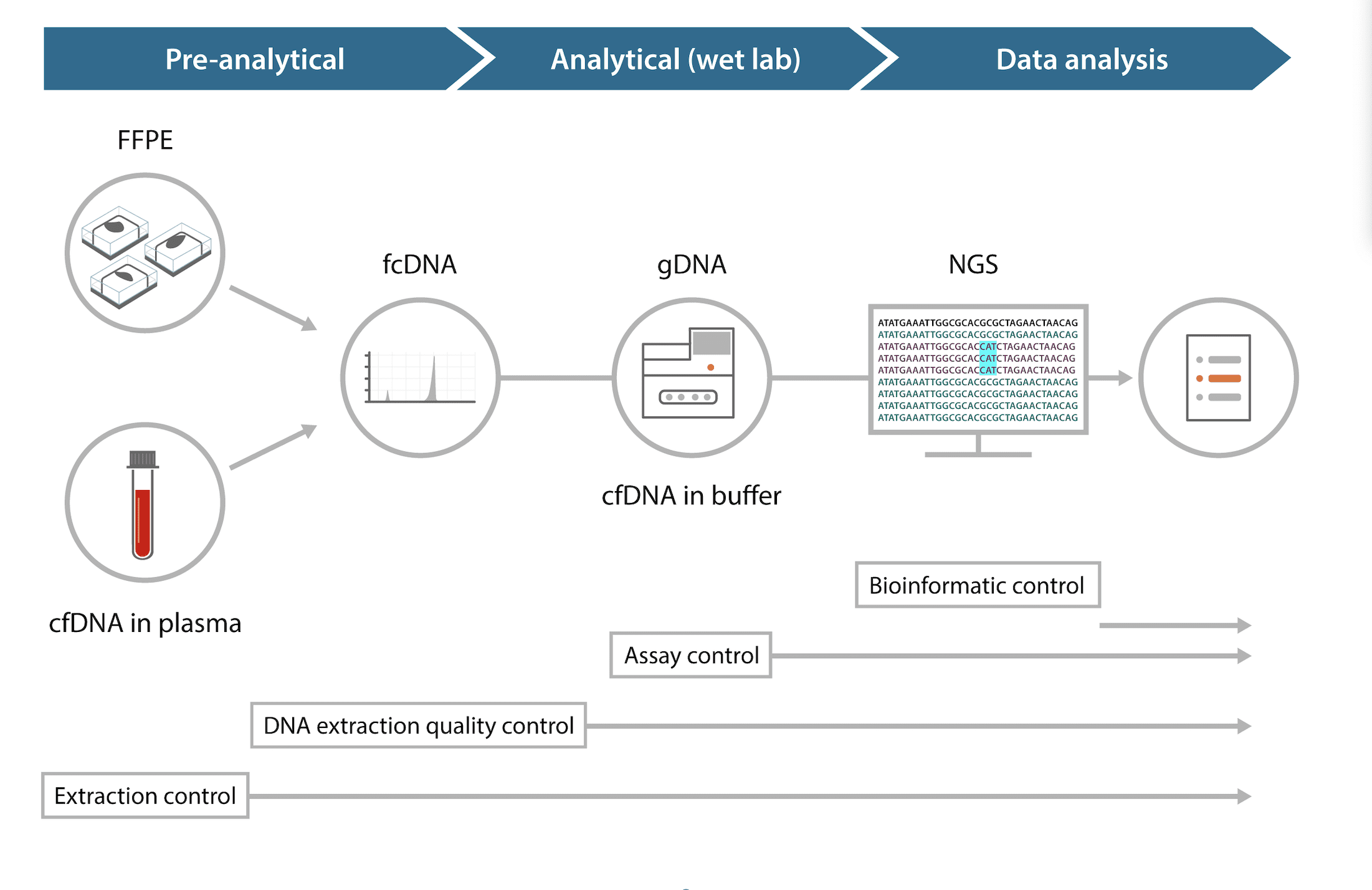
Molecular assays in oncology, including NGS, involve multiple steps, which are prone to biological and technical errors (Figure 1). The preanalytical workflow spans sample collection, processing, storage, and transportation, all of which are critical in maintaining the quality and purity of samples. For solid tumor biopsies—the gold standard in tumor characterization—sample collection is performed via various surgical procedures, followed by preserving the tissue by flash freezing or with chemicals and slicing the samples into thin sections. However, the lack of standardization in specimen processing typically leads to inefficient and poor reproducibility of results. Formalin fixation of tissues damages nucleic acids, leading to adverse effects on downstream molecular processes. Formalin-fixed, paraffin-embedded (FFPE) samples display much lower quality of NGS reads compared to matched fresh frozen tissues.1 The major sequence artifacts in FFPE samples include chimeric reads (noncontiguous sequences) arising from fragmentation, abasic sites, and base modifications caused by cytosine deamination.2
A more recently adopted noninvasive method of sample collection—liquid biopsy—also suffers from a lack of standardization. Liquid biopsy is used for analyzing circulating tumor DNA (ctDNA), also known as cell-free DNA (cfDNA), in peripheral blood drawn from patient samples. Specialized tubes for blood collection prevent lysis of white blood cells to prevent genomic DNA from contaminating the cfDNA. However, there could be variations in the capabilities of different tubes in preserving samples. Once the samples are collected, appropriate storage conditions should be maintained until sample processing to prevent DNA denaturation over time. Harmonization of the preanalytical processes in molecular diagnostics is thus essential in obtaining high-quality, reproducible results.
Following preanalytical workflows, analytical methods in genetic profiling consist of technologies that either analyze a single gene or variant, or multiple genes and variants. For any test result to be useful, it is imperative to identify variabilities in the process and control it by employing appropriate quality assessment measures.
Molecular processes that study DNA from limited samples use PCR amplification steps to increase DNA quantity. Library preparation in NGS also involves multiple rounds of PCR amplification, including the adaptor ligation step. Artifacts introduced by PCR and other sequencing chemistries add to the already existing artifacts introduced by preanalytical factors and can limit interpretation of results. Errors during sequencing are relatively well understood and are controlled by an automated internal error rate calculation in the sequencer known as Phred quality score.
However, after the data is obtained from sequencing there is a high degree of variability in variant calling due to insufficient standardization of bioinformatics pipelines. Processing raw data to detect specific variants comprises multiple steps that are generally platform-specific and use different software to assess raw data quality and filter it, align the sequence with a reference genome, and identify and annotate variants. The robustness of data analysis can be technically challenging in calling out low variant allele frequencies and structural variants, so choosing the right algorithm is crucial in calling out different types of variants. In silico data sets with known variants and allele frequencies can serve as quality control tools for standardization of bioinformatics pipelines.
Monitoring Quality and Mitigating Errors
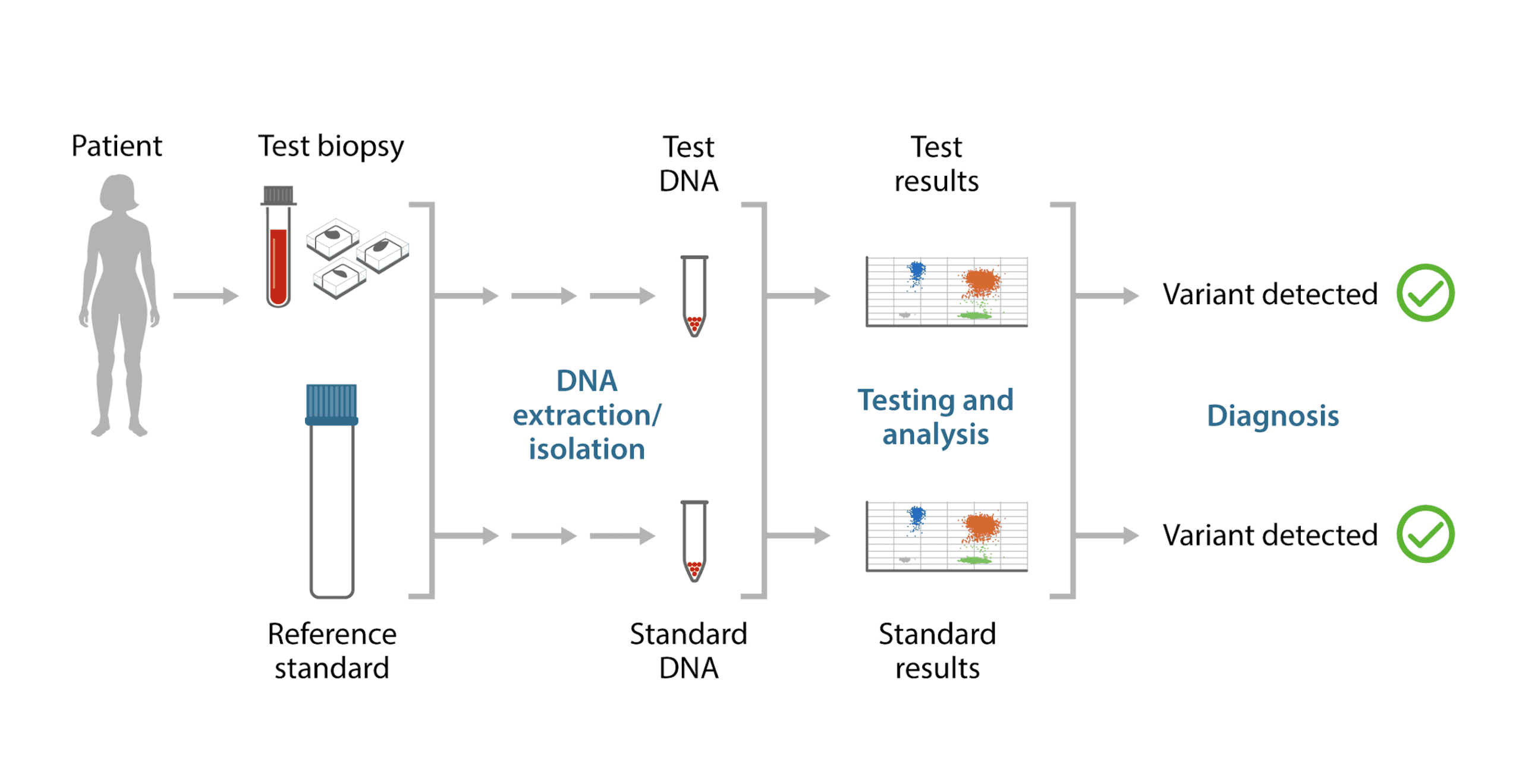
Based on these premises, strong quality control measures are required in each step of the workflow to identify, monitor, and mitigate errors. The quality of samples, input quantity, and fragment length of library are among the major factors that need to be tracked and controlled along the NGS workflow to consistently achieve good sequencing runs. Appropriate reference materials formulated to mimic patient samples serve as good quality controls by acting as performance indicators in the workflow. There are published guidelines for NGS-based cancer diagnostic tests by various regulatory bodies that establish the validation requirements for implementation of these tests in clinical and public health laboratories. Recommendations include the use of positive, negative, and sensitivity controls in the NGS workflow.3 The chosen reference material should ideally mimic patient samples and be used for calibrating the preanalytical processes in determination of quality and quantity of analytes.
The preanalytical controls should aid in assessing quantification and purity determination of samples. A good quality DNA sample has an OD260/280 ratio of 1.8 to 2.0, and a pure sample devoid of contamination should have an OD260/230 ratio of 2.0 to 2.2. The input quantity required will depend on the NGS library preparation kit and sample type. The appropriate method of quantitation will depend on the sample type; however, fluorometric-based methods are recommended.
For FFPE and other damaged samples, an initial DNA restoration step, involving a commercially available kit, can be used to repair some common errors found in FFPE samples like nicks and gaps, blocked 3’ ends, oxidized bases, and deamination of cytosine to uracil. This DNA repair step helps in discerning true mutations from sequence artifacts caused by DNA lesions in FFPE samples. The quality of FFPE DNA can be evaluated by various capillary gel electrophoresis methods and qPCR methods that determine the amplifiability of DNA. In liquid biopsy samples, the presence of high molecular weight fragments in the capillary gel electrophoresis shows the presence of contaminating genomic DNA, whereas the presence of shorter DNA fragments in FFPE samples indicates higher degradation.
In the analytical step, it is recommended to choose controls with multiple known variants at low allele frequencies to establish and verify assay sensitivity. The New York State Department of Health guidelines recommend validation of performance characteristics for each variant separately, and for each format type of patient sample, without sample type bias.3 Key performance parameters include analytical specificity and sensitivity, robustness, and reproducibility and limit of detection. The analytical sensitivity of an NGS assay is the ability to correctly detect true positives especially at low allele frequencies. The specificity of the assay is an indicator of the precision of detection which is measured by not detecting false positives.
In the postanalytical step, the raw data generated by different sequencing platforms can vary depending on the sequencing chemistry used. Processing of raw data into annotated data requires various software programs and tools, and the variant detection could largely depend on the kind of software used in the pipeline. An in silico data set control with verified variants at various allele frequencies can help in establishing robustness and precision of the data analysis pipeline without software bias.
Controls for Cancer Diagnostics
Reference standards allow evaluation of a given assay method by checking for consistencies with the expected values of a defined parameter in the control material. However, it is important to ensure commutability of controls with patient samples. Cell line-derived reference standards offer the genomic complexity of patient samples, whereas synthetic genes provide minimal genomic complexity. Patient samples relevant to specific disease or cancer types may be difficult to acquire routinely. Reference standard materials derived from cell lines offer reproducible, renewable, and affordable experimental controls for molecular diagnostic assays. Cell line-derived reference standards have evolved from single gene pairs of wild type and corresponding mutant gene variants to multiplexed cocktails of a larger number of genes and variants implicated in cancers. These large gene panels are especially useful to identify new biomarkers linked to uncontrolled cell growth of cancers, and to better characterize genes known to affect cancer growth.

When striving for full assay control to monitor workflows from DNA extraction to assay results, reference material in FFPE format offers a complete solution (Figure 2). One such example of a cell line-derived reference material is OncoSpan FFPE (Horizon Discovery), which offers over 370 variants across 150 genes associated with cancer and variant allele frequencies (VAFs) spanning 1% to 95% to aid in calibrating assays with a varied range of detection sensitivities. Reference standards that include orthogonal validation by droplet digital PCR (ddPCR) on a subset of mutations offer a two-layer verification of the claimed variants.
Flexible reference materials that are well suited for use with a large set of gene panels enable researchers to assess assay suitability across a wide range of conditions. Accompanying batch-specific in silico whole exome sequencing data allows for further monitoring and troubleshooting bioinformatics alignment. The added benefit of sequence data provides more in-depth analysis of the genes and variants linked to a particular disease.
On the Horizon

NGS assay development shall continue to expand in the medical community and provide significant benefits to patient care. In doing so, NGS assays will become more mainstream in their use but also require appropriate controls to monitor assay activity. Cell line-derived reference materials provide a highly useful, renewable, and reproducible source of relevant genes and associated VAFs for ongoing cancer research and detection.
Keith Cannon, MBA, is director of commercial product management, diagnostics at PerkinElmer’s Horizon Discovery business, a global leader in the application of gene editing and gene modulation for cell line engineering. He earned his MBA from Johns Hopkins. Before joining Horizon, Cannon led efforts at Aviva Biosciences to develop and commercialize methods to recover and enrich rare cells for liquid biopsy. Presently, Cannon is responsible for portfolio management and sales for reference standards at Horizon Discovery.
Prabha Nagarajan, PhD, is a senior research scientist in the diagnostics team of PerkinElmer’s Horizon Discovery business, UK. She obtained her PhD from University of Rochester where she studied mitochondrial DNA repair mechanisms. Before joining Horizon Discovery, she worked in the clinical diagnostics team at Strand Life Sciences in India. Her present role at Horizon involves developing diagnostic reference standards for use in oncology and other genetic profiling tests.
References
1. Spencer DH, Sehn JK, Abel HJ, Watson MA, Pfeifer JD, Duncavage EJ. Comparison of clinical targeted next-generation sequence data from formalin-fixed and fresh-frozen tissue specimens. J Mol Diagn. 2013;15(5):623-633. doi:10.1016/j.jmoldx.2013.05.004.
2. Do H, Dobrovic A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clin Chem. 2015 Jan;61(1):64-71. Epub 2014 Nov 24. PMID: 25421801. doi: 10.1373/clinchem.2014.223040.
3. New York State department of Health. Oncology – Molecular and Cellular Tumor Markers. Next Generation” Sequencing (NGS) guidelines for somatic genetic variant detection. March 2015. Available at: https://www.wadsworth.org/sites/default/files/WebDoc/1300145166/NextGenSeq_ONCO_Guidelines.pdf. Accessed October 2, 2020.

{kind=link}