A novel statistical approach facilitates classification of variants in high- and moderate-risk genes
By Karla R. Bowles, Brian Morris, Krystal Brown, Elisha Hughes

Karla R. Bowles, PhD, FACMG, Myriad Genetic Laboratories.
Over the past couple of decades, genetic testing has evolved from targeted gene testing for a limited number of genetic diseases to analysis of large gene panels or whole-exome sequencing for a multitude of clinical conditions. The use of next-generation sequencing (NGS) technologies for large-panel or whole-exome analysis generates vast amounts of DNA sequence data.
While it is possible to estimate the cancer risks associated with many DNA variants identified during such testing, often variants of uncertain significance (VUS) are also identified. These variants represent a challenge for healthcare providers and patients, as they may create uncertainty about an individual’s diagnosis and appropriate clinical management. Traditional approaches to variant classification are often not suitable for reclassifying VUS as either pathogenic or benign. This is especially true for genes associated with moderate disease risks, for which less information of relevance may be available.

Brian Morris, Myriad Genetic Laboratories.
This article describes our development of a statistical approach, called a “history weighting algorithm” (HWA), for reclassifying variants identified in genes associated with significant risk of breast cancer—specifically, ATM, BRCA1, BRCA2, CHEK2, and PALB2. This first-in-class tool measures the severity of personal and family cancer histories associated with a specific variant in order to classify the variant as pathogenic or benign. It is a highly accurate and objective classification tool, with positive and negative predictive values >0.995. Importantly, it overcomes many of the limitations of traditional classification tools, such as segregation and functional analyses, which require additional testing of multiple family members and biochemical assays, respectively.
While the HWA tool was initially developed for analyzing variants identified in high-risk breast cancer genes (BRCA1, BRCA2), its utility for analyzing variants in moderate-risk genes (ATM, CHEK2, PALB2) makes HWA a groundbreaking tool. We believe that with additional development and modification, the HWA tool could be adapted for classifying variants identified in additional autosomal-dominant genes associated with other genetic conditions.
BACKGROUND
Genetic testing using NGS gene panels has become common clinical practice for assessing an individual’s hereditary cancer risk. In accord with guidelines issued by professional societies in the field, the identification of disease-causing genetic variants during clinical genetic testing may influence an individual’s medical management.1,2 Hereditary breast and ovarian cancer syndrome (HBOC), for example, is associated with the BRCA1 and BRCA2 genes. Pathogenic variants in these genes are associated with a lifetime risk for breast cancer of up to 87%, and for ovarian cancer of up to 44%. These significant cancer risks have resulted in the issuance of National Comprehensive Cancer Network (NCCN) recommendations that women with a pathogenic variant in BRCA1 or BRCA2 receive increased screening, and consider risk-reducing surgery, such as mastectomy or salpingo-oophorectomy.1

Krystal Brown, PhD, Myriad Genetic Laboratories.
Given the medical management implications of pathogenic variants in cancer-predisposition genes, accurate variant classification is a vital component of genetic testing. Multiple lines of evidence are required to assign a clinical significance (pathogenic or benign) to a variant.3,4 Pertinent evidence to be considered may include direct evidence that the variant changes the structure or function of a gene as well as clinical evidence that the variant is segregated in families with a high incidence of cancer.
For many genetic variants, however, there is insufficient evidence available to determine their clinical significance. Such VUS are typically rare and often represent single amino acid changes, the insertion or deletion of a small number of amino acids, or changes in intronic sequences. Other variant types may also be classified as VUS.
Although identification of a VUS in a breast cancer predisposition gene is not by itself sufficient grounds for medical intervention, the reporting of a VUS can cause uncertainty and anxiety about medical management and cancer risk. In addition, appropriate medical care may be delayed for individuals carrying a VUS that is later determined to be pathogenic.
For many VUS, direct evidence of changes to gene function may not be readily acquired. Using current classification methods, the process of amassing enough data to evaluate clinical significance can take years, or even decades.
Historically, genetic testing has been limited to such high-penetrance genes as BRCA1 and BRCA2. Data available from more than 20 years of genetic testing has resulted in as few as 2% of patients tested for BRCA1 and BRCA2 receiving a reported VUS within these genes.3 However, many moderate-risk genes are now also known to be associated with increased breast cancer risk. Such moderate-risk genes include ATM, CHEK2, and PALB2, which are associated with lifetime breast cancer risks ranging from 17% to 58%.5–7

Elisha Hughes, PhD, Myriad Genetic Laboratories.
Unfortunately, the clinical significance of genetic variants in such moderate-risk genes can be more difficult to evaluate than in high-risk genes. Such increased difficulty arises mostly from two limitations: clinical testing for most moderate-risk genes has only been available for about 3 years, and the lower incidence of cancer resulting from moderate-risk genes inherently limits the amount of clinical information available. Ultimately, these factors reduce the amount of data available for evaluating the clinical significance of variants in moderate-risk genes, which in turn results in a higher incidence of VUS. With the expansion of genetic testing to pan-cancer panels that include such moderate-risk genes, it is vitally important that laboratories correspondingly increase the capabilities of the tools available for accurate and timely variant classification.
HISTORY WEIGHTING ALGORITHM
Recently, a history weighting algorithm (HWA) was developed to determine the clinical significance of BRCA1 and BRCA2 genetic variants.8 This novel method is based on the premise that personal and family cancer history should be related to the clinical significance of the variant. Specifically, pathogenic variants will be identified more frequently among individuals with a strong personal and family history of gene-associated cancers (breast and ovarian cancer in the case of BRCA1 and BRCA2). Conversely, benign variants should be identified independent of cancer history.
HWA is a powerful variant classification method, as it uses statistical analysis of clinical data for individuals with a specific VUS to identify trends in personal and family cancer history that allow the clinical significance of the variant to be evaluated. This method is able to capitalize on the recent uptake of clinical genetic testing to accelerate data acquisition relative to traditional classification methods. In order to aid in variant classification for moderate-risk genes, we describe here the development of an HWA for ATM, CHEK2, and PALB2.
HWA DEVELOPMENT
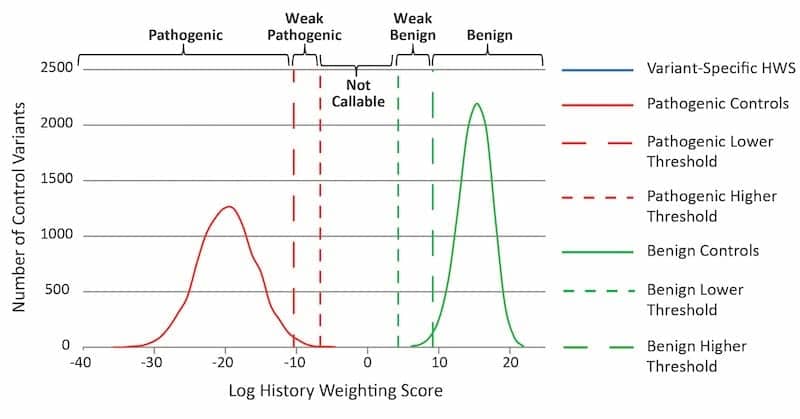
Figure 1. Illustration of an HWA graph. The variant-specific HWS is compared to those of 10,000 pathogenic and 10,000 benign composite control variants. Variant classification categories (top) are defined by thresholds based on composite control HWS distributions. Click to expand.
All individuals included underwent clinical genetic testing with a 25-gene hereditary cancer panel that included sequencing and large-rearrangement analysis of ATM, CHEK2, and PALB2. Clinical information for tested individuals was obtained from provider-completed test request forms. Such information encompassed age at testing, ancestry, and personal and family cancer history, including cancer type(s) and age(s) of diagnosis, if applicable. Individuals were excluded from analysis if their personal or family history was not provided or if the individual was known to carry a pathogenic variant or VUS in BRCA1, BRCA2, ATM, CHEK2, or PALB2, in addition to the variant being analyzed.
In order for the HWA to evaluate a specific variant, a history weighting score (HWS) was first calculated for the variant. Based on the severity of personal and family cancer history, a statistical weight was assigned to each proband carrying the specific variant. These weights were combined to give a final HWS for that variant. The clinical significance of each specified variant was evaluated by comparing the variant-specific log HWS against HWS distributions composed of 10,000 known pathogenic variants (positive control distribution) and 10,000 benign variants (negative control distribution) in the same gene (see Figure 1).
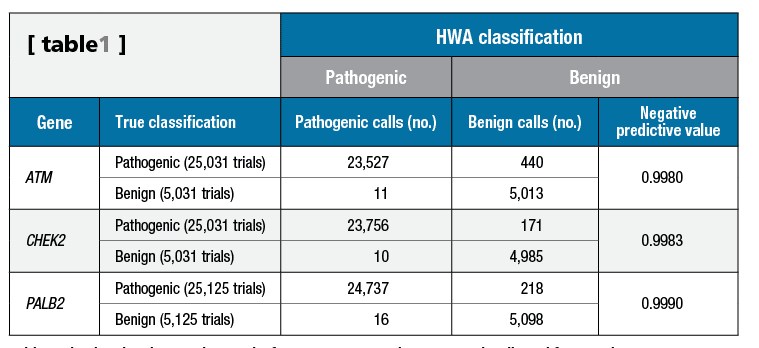
Table 1. Simulated variant testing results for ATM, CHEK2, and PALB2. NPV is adjusted for prevalence. Click to expand.
During HWA development, more than 30,000 simulated pathogenic and benign variants were constructed utilizing proband data from 119,452 patients tested using a 25-gene pan-cancer panel that included ATM, CHEK2, and PALB2. HWS classification thresholds were established for the positive and negative control distributions, and used to determine whether a variant would most likely be benign. These thresholds were calculated to yield negative predictive values >0.995 (see Table 1).
CLASSIFICATION THRESHOLDS
The accuracy of the HWA was dependent on the establishment of classification thresholds, which were utilized by the calculation to call a variant benign. For a VUS to be called benign, the HWS had to be greater than the 99.5th percentile plus a gene-specific number of standard deviations of the positive control HWS distribution, and greater than the 1st percentile of the negative control HWS distribution (see Figure 2). This calculation represents a high degree of certainty that the personal and family histories associated with the variant are both different from those associated with pathogenic variants in the same gene, and similar to those of benign variants in the same gene.

Figure 2. HWA graphs illustrating classification calls for select variants in ATM, CHEK2, and PALB2. Click to expand.
The thresholds established for the HWS relative to the positive and negative control HWS distributions were established using more than 30,000 simulated pathogenic and benign variants with a resultant negative predictive value ?0.9980 (see Table 1). Positive predictive value was not calculated, as the HWA is not currently designed to upgrade variants in moderate-risk genes from VUS to pathogenic or likely pathogenic.
DISCUSSION
Determining the clinical significance of a VUS in a timely and accurate manner is critical for patient care. While a great deal of information is available for high-risk genes such as BRCA1 and BRCA2, there is less functional and clinical data available for most moderate-risk genes.
As illustrated by the work described here for ATM, CHEK2, and PALB2, the HWA provides a robust, accurate, and quantitative tool to determine the clinical significance of VUS in these genes. The method adds to traditional classification tools, and is even more important in light of the shift to pan-cancer panel testing that includes both high- and moderate-risk genes.
Many limitations of traditional variant classification techniques are addressed by the HWA method. The HWA does not rely on testing multiple relatives from large pedigrees, which is required for segregation analysis. Additionally, the HWA is a more direct measure of a variant’s clinical significance than classification techniques that measure mRNA splicing or protein function. For such methods, the functional effect measured in a laboratory may not directly relate to clinical outcomes. In addition, it is often unclear how to interpret results showing partial, but not complete, loss of function. By contrast, the HWA more directly measures a variant’s clinical impact by determining its relationship to relevant personal and family cancer histories.
CONCLUSION
We developed and implemented the HWA to aid in the reclassification of VUS in the moderate-risk breast cancer genes, ATM, CHEK2, and PALB2, to a definitive, benign classification. Such reclassification enables improved patient care and decreased uncertainty and anxiety regarding genetic testing results.
The high accuracy of the HWA makes this classification technique the gold standard for reclassification of ATM, CHEK2, and PALB2 VUS in the clinical diagnostic setting. Additional modifications of the HWA may allow this tool to be extended to autosomal dominant genes associated with clinical conditions other than cancer.
Karla R. Bowles, PhD, FACMG, is senior laboratory director; Brian Morris is a software engineer; Krystal Brown, PhD, is a technical writer; and Elisha Hughes, PhD, is a research biostatistician, at Myriad Genetic Laboratories Inc. For further information, contact CLP chief editor Steve Halasey via [email protected].
REFERENCES
- Daly MB, Pilarski R, Axilbund JE, et al. Genetic/familial high-risk assessment: breast and ovarian, version 2.2016. NCCN Clinical Practice Guidelines in Oncology. Fort Washington, Pa: National Comprehensive Cancer Network, 2016. Available at: www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Accessed March 3, 2016.
- Provenzale D, Gupta S, Ahnen DJ, et al. Genetic/familial high-risk assessment: colorectal, version 1.2016. NCCN Clinical Practice Guidelines in Oncology. Fort Washington, Pa: National Comprehensive Cancer Network, 2016. Available at: www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf. Accessed July 19, 2016.
- Eggington JM, Bowles KR, Moyes K, et al. A comprehensive laboratory-based program for classification of variants of uncertain significance in hereditary cancer genes. Clin Genet. 2014;86(3):229–237; doi: 10.1111/cge.12315.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424; doi: 10.1038/gim.2015.30.
- Cybulski C, Wokolorczyk D, Jakubowska A, et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J Clin Oncol. 2011;29(28):3747–3752; doi: 10.1200/jco.2010.34.0778.
- Antoniou AC, Casadei S, Heikkinen T, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371(6):497–506; doi: 10.1056/nejmoa1400382.
- Ahmed M, Rahman N. ATM and breast cancer susceptibility. Oncogene. 2006;25(43):5906–5911; doi: 10.1038/sj.onc.1209873.
- Pruss D, Morris B, Hughes E, et al. Development and validation of a new algorithm for the reclassification of genetic variants identified in the BRCA1 and BRCA2 genes. Breast Cancer Res Treat. 2014;147(1):119–132; doi: 10.1007/s10549-014-3065-9.



{kind=link}