One-sentence summary:
Researchers at UCLA identified DNA methylation entropy as a new and accurate biomarker for aging, offering deeper insight into epigenetic changes over time.
Three brief takeaways:
- Methylation entropy reflects the randomness in DNA chemical tags and changes predictably with age.
- It predicts biological age as accurately as traditional methods and improves further when combined with other markers.
- The study supports the idea that loss of epigenetic information plays a key role in aging and may guide future anti-aging therapies.
A new research paper was published in Aging (Aging-US) Volume 17, Issue 3, titled “DNA methylation entropy is a biomarker for aging.”
Researchers Jonathan Chan, Liudmilla Rubbi, and Matteo Pellegrini from the University of California, Los Angeles, led a study that discovered a new way to measure changes in DNA that can help predict a person’s age. This method focuses on how random certain chemical tags on DNA become over time. The team compared this new measurement, called methylation entropy, to existing methods and found it performed just as well—or even better. These findings support the idea that changes in our epigenetic information are closely linked to aging and could offer new tools for studying age-related diseases.
DNA Methylation Turns Genes ‘On’ and ‘Off’
The study focused on DNA methylation, a process where chemical marks are added to DNA and help control which genes are turned on or off. Scientists have traditionally measured average methylation levels to estimate biological age using “epigenetic clocks.” This study, however, takes a different approach. The researchers used buccal swabs (cells from inside the cheek) from 100 individuals between ages 7 and 84 and applied targeted bisulfite sequencing techniques to measure methylation entropy across 3,000 regions of the genome.
The Entropy Factor
Entropy in this context reflects how disordered or varied the methylation patterns are at certain sites on the DNA. The researchers discovered that as people age, the entropy of methylation at many locations changes in a reproducible way. Sometimes it increases, reflecting more random patterns, and sometimes it decreases, showing more uniformity. These shifts are not always tied to how much methylation is happening, which suggests entropy provides new information beyond what traditional methods can offer.
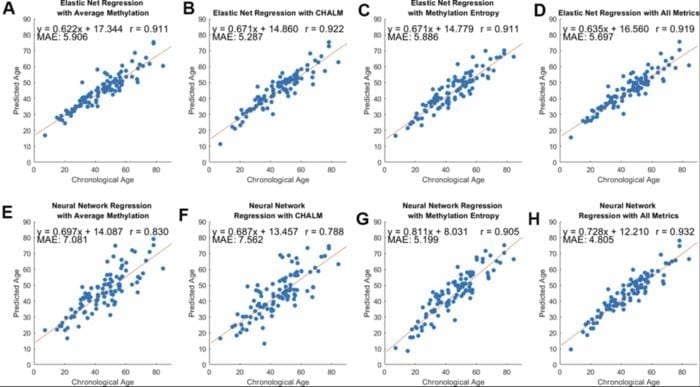
To test how well this new metric could predict age, the team used both statistical and machine learning models. They found that methylation entropy predicted age as accurately as traditional methods, and the best results came from combining entropy with other measurements like average methylation and a method called CHALM. These combined models were able to estimate age with an average error of just five years.
This research supports the growing theory that aging is partly caused by a gradual loss of epigenetic information—the biological “instructions” that help keep our cells working properly. This insight also connects with recent studies suggesting that restoring this lost information might reverse some signs of aging. While more research is needed to study methylation entropy in other tissues, this work points to a more precise and powerful way to measure biological aging, which could influence the future of aging-related treatments and therapies.
Featured Image: Model performance across three methylation metrics and two regression methods. Pearson’s correlation coefficient, equation of best-fit line, and MAE between predicted and chronological age are included. (A–D) Predicted versus chronological age using average methylation, CHALM, entropy, and a combination of these metrics with elastic net regression. (E–H) Predicted versus chronological age with neural network regression. Image: Copyright: © 2025 Chan et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.




{kind=link}