
Advanced technologies can streamline assay workflows in research and clinical environments
By Richard A. Montagna, PhD, FACB
In the United States, diagnostic tests generally arrive at the laboratories where they’re performed by following one of two paths. Manufacturers of in vitro diagnostics (IVDs) introduce tests onto the market by demonstrating their safety and efficacy to FDA, and gaining the agency’s clearance or approval for market entry. Meanwhile, many laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA) take an alternative path, whereby they develop and validate their own “laboratory-developed tests” (LDTs). This practice is consistent with FDA’s definition of an LDT as a diagnostic test that is “developed, validated, and performed in an individual laboratory.”1

Richard A. Montagna, PhD, FACB, Rheonix Inc.
Among both FDA-cleared tests and LDTs, an increasing proportion of marketed tests are molecular diagnostics that are building upon the wealth of genetic information gradually being gathered through research into the genomes of humans and infectious agents. Such genomic information enables rapid and sensitive detection of infectious agents, characterization of malignant tumors, prediction of sensitivity to pharmacologic agents, and stratification of oncology patients into responders and nonresponders with regard to specific therapeutic interventions.2–5 Moreover, analysis of an individual’s DNA can provide valuable information about their possible future health risks.6 Taken together, these capabilities enable healthcare professionals to make better informed and personalized decisions about appropriate medical treatment for their patients.
Most molecular diagnostics rely upon gene amplification technologies to copy the genetic targets of interest, so that they can be detected against a background of other genetic information. Several alternative gene amplification methods are now available to exploit the growing base of DNA sequence data. Nevertheless, polymerase chain reaction (PCR)—the earliest method of gene amplification—continues to dominate the landscape of tests intended to detect specific gene sequences, both for IVDs and LDTs. The proliferation of such PCR-based testing has led to the development of a variety of commercial platforms designed to simplify and automate the use of PCR in both clinical and nonclinical applications.

Figure 1. The Rheonix CARD cartridge is an inexpensive, injection molded microfluidic device that contains all the pumps, valves, and reagent and reaction reservoirs needed to perform complex molecular assays.
Regardless of application, the amplified products of PCR are generally detected using either “real-time” fluorescence methods or “endpoint” detection of amplicons on DNA arrays. Each approach has its own distinct advantages. While real-time methods can provide accurate quantitative results, the total number of targets is often limited by the number of fluorescent dyes available. By contrast, endpoint detection methods that utilize DNA arrays to detect PCR amplicons at the completion of an assay can detect a higher number of targets, but can generally provide only semiquantitative data about the concentration of the targets. In settings where such semiquantitative data are sufficient, endpoint detection methods provide a powerful means to perform highly multiplexed PCR assays.
In clinical laboratory settings, automated PCR systems prove their value by being able to achieve true “sample-to-actionable-results,” with little or no intervention by laboratory personnel. Ideally, such automation should encompass all sample preparation steps, including DNA or RNA isolation, multiplexed PCR, and detection. But to improve their chances of adoption in clinical settings, such systems should go beyond mere automated ease of use to address other issues common in most clinical labs, including space limitations, the capital cost of instrument acquisition, and the cost per test for any necessary disposables.
A cogent example is provided by the recent decision of the Centers for Medicare & Medicaid Services (CMS) to extend its timeline for implementing the new market-based payment system for clinical labs, pursuant to the Protecting Access to Medicare Act of 2014.7 Despite this temporary reprieve, concerns about the cost per test rank high in the minds of clinical laboratory directors facing the possibility of declining reimbursements in the not-too-distant future.
THE RHEONIX PLATFORM
With an eye toward addressing the diverse concerns of clinical laboratory professionals, Rheonix Inc, Ithaca, NY, has developed a fully automated molecular diagnostic system designed to live within existing space constraints, streamline laboratory workflow, and achieve a favorable and sustainable cost structure. The platform consists of the disposable Rheonix Chemistry and Reagent Device (CARD) microfluidic cartridge (see Figure 1), and the Encompass Optimum workstation (see Figure 2).

Figure 2. The Encompass Optimum workstation can perform complex molecular assays, and provides true “sample in–results out” capabilities by automatically processing up to 24 individual specimens at a time.
The CARD cartridge contains all of the pumps, valves, reagent and reaction reservoirs, and connecting microchannels necessary to achieve seamless automation. The unique design of the pumps and valves allows bidirectional microfluidic flow, thus providing a means to effectively and easily achieve reagent mixing and facilitate such assay steps as cell or viral lysis, and DNA or RNA extraction. In addition, the CARD cartridge also contains onboard waste storage so that at the end of any assay, it can be treated as biohazard waste and safely discarded.
The workstation’s intuitive software controls the robotic system that delivers samples and reagents to the CARD cartridge as needed, provides the systematic pneumatic signals required to move various fluids throughout the microfluidic device, and finally interprets and reports the assay results. By means of the workstation’s touchscreen and user interface, a user is walked through the various initial steps required to load the sample rack and disposables, including the CARD cartridges and reagent pack. Then, once the system is readied, a simple touch of the “go” button on the touchscreen will initiate the totally automated process. Together, the workstation and a total of six CARD cartridges—each one capable of processing four separate samples—can automatically process up to 24 individual specimens within a 3 to 4 hour period.
USING THE CARTRIDGE AND WORKSTATION
The Rheonix platform does not require operators to perform any preliminary treatment of samples. “Raw” samples can be loaded directly into the cartridge for automatic analysis. A broad spectrum of sample types have been successfully analyzed, including buccal swabs, endocervical swabs, plasma, serum, urine, vaginal swabs, and whole blood, as well as fresh and formalin-fixed, paraffin embedded (FFPE) tissue blocks (For more information, see “Streamlining Library Preparation for Next-Generation Sequencing“).
Once the samples and consumables are loaded into the cartridge, the automated assay routine will lyse cells, isolate and purify nucleic acids (either RNA or DNA), perform PCR to amplify the multiplex of desired targets, and deliver the amplicons to the DNA array. There, the hybridization products are detected by the workstation’s optics, and the presence or absence of the defined targets of interest is then reported by the workstation’s software.
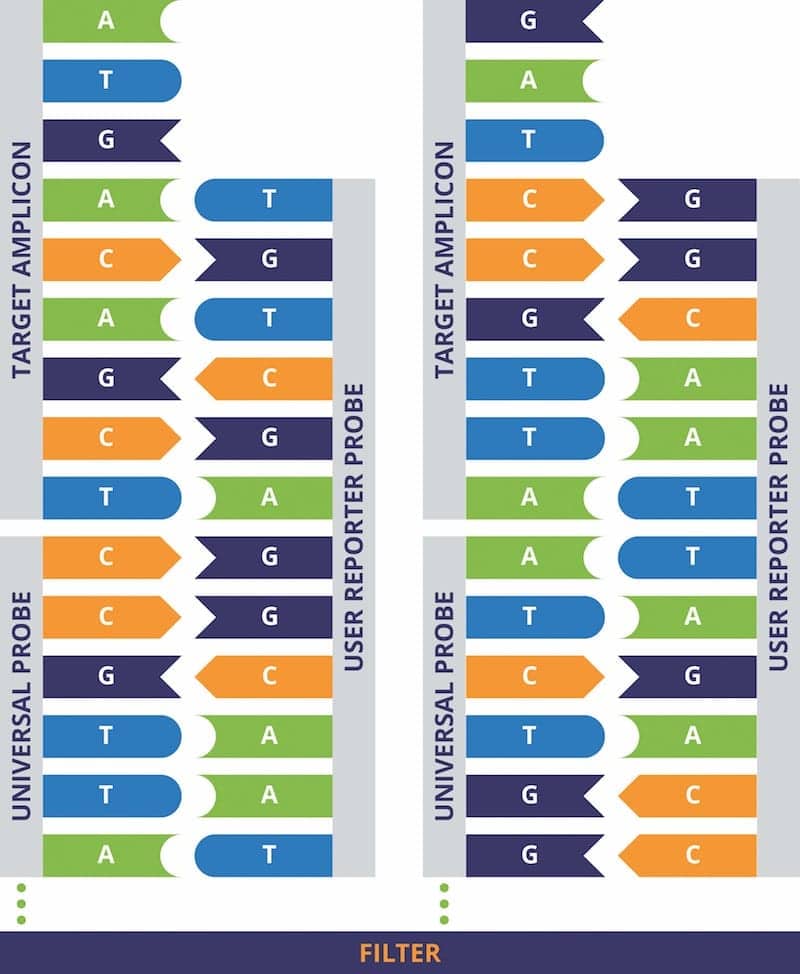
Figure 3. Use of universal probes to detect specific PCR amplicons. The DNA array contained within the Universal CARD cartridge contains 20 different “universal probes” immobilized at specific locations on the array (two are shown). The universal probes are immobilized onto the array via linkages through their 5′ termini. User-defined “reporter” probes are introduced; after they have hybridized to the immobilized universal probes, their 5′ sequences are available to hybridize to complementary sequences found in the PCR amplicons. Click to expand.
To enable users to achieve automatic functionalization of their DNA arrays, Rheonix has developed a Universal CARD cartridge that has a lawn of 20 different “universal” probes immobilized at defined locations on the array via their 5′ termini. These universal probes are not related to any particular DNA targets, but serve as a means to “anchor” a user’s specific reporter probes designed to hybridize to PCR amplicons. The reporter probes hybridize to the universal probes via complementary sequences contained within the reporter probes’ 3′ termini. The remaining sequences within the 5′ portion of the user’s reporter probes are then available to hybridize to the targets of interest (see Figure 3).
The workstation enables users to automatically functionalize DNA arrays during the development of their own unique assays. The system’s ability to easily functionalize DNA arrays makes it possible for users to evaluate and optimize their own reporter probes and hybridization conditions “on the fly,” thereby simplifying assay development.
To ensure that the integrated optical workstation and software can detect and correctly interpret the presence or absence of specific PCR amplicons, it is essential that the specified DNA probes be localized in defined positions on the DNA arrays. The system’s time-to-results can be further streamlined by taking advantage of the workstation’s ability to simultaneously perform multiple functions in parallel, allowing DNA arrays to be automatically functionalized while the sample preparation and multiplex PCR steps are already under way. In this way, no additional time is consumed in preparing the DNA arrays. When the multiplex PCR reactions are completed, the DNA arrays should already be available to hybridize to the PCR-generated amplicons.
Once an array is finalized, the user-defined reporter probes hybridized to the PCR amplicons can be detected and reported by the workstation’s optics and integrated software. This Universal CARD approach has been successfully used to detect a variety of infectious agents and single nucleotide polymorphisms.
Users can control all aspects of the workstation’s automated processes in order to design, optimize, and ultimately finalize their own unique assays. In this way, users can optimize the DNA isolation process, delivery of required volumes of reagents, mixing rates and resident times in the various CARD cartridge reservoirs, PCR thermocycling conditions (ie, cycling times and temperatures), and the functionalization of the terminal DNA arrays. The workstation aids the user by providing default settings for such parameters, but all functions can be adjusted as needed. Once the user’s assay parameters have been optimized and the information stored in the workstation’s software, the workstation will run the finalized assay routine without any user intervention.
THE PLATFORM IN REGULATORY CONTEXT
Rheonix has directed a significant measure of its design efforts for the Universal CARD cartridge and the Encompass Optimum workstation toward meeting the needs of CLIA-certified laboratories seeking to develop their own, in-house tests. The result is a versatile platform that simplifies many of the steps required to create a customized test, and automates test performance as well as the reporting of test results.
At the same time, the company has continually monitored the progress of FDA’s proposed framework for the regulation of LDTs, and has frequently spoken with FDA officials in order to gain insight into the agency’s thinking about this controversial area. FDA’s draft guidance documents on the regulation of LDTs were released about 2 years ago, and have provided useful guidance for development of the Rheonix platform.1,8 Equally important, early discussions with the agency have reinforced the company’s strategy of creating a versatile platform that will enable labs to design their own LDTs while remaining compliant with potential changes in the regulatory landscape.
Rheonix discussions with FDA have also explored the agency’s requirements for obtaining clearance of a “dual use” workstation that can perform either FDA-cleared assays or user-defined assays (but not both at the same time). The agency has recommended that Rheonix design a clinical study to gather objective evidence that the Encompass Optimum workstation is capable of toggling between performing FDA-cleared and user-defined assays without negatively affecting the performance of the cleared assays (For more information, see “‘Dual Use’ Assay Capabilities“).
Toward that end, the company plans to initiate such a study in early 2017. Once cleared for “dual use,” the workstation will offer a versatile platform of great value to clinical laboratories seeking to develop user-defined assays. Presently, the Encompass Optimum workstation is available for research use only.
CONCLUSION
FDA and CMS are continuing to engage in vigorous discussions with IVD manufacturers and the major clinical laboratory associations and professional societies over the final shape of regulations for LDTs. A number of alternatives to FDA’s proposed regulatory framework have been suggested, so it is by no means certain that the agency’s approach will prevail.
Meanwhile, Rheonix is staying abreast of potential regulatory changes that might affect its ability to provide clinical laboratories with a robust, automated system that will meet their needs to create customized in-house tests while remaining compliant with new regulations. Whatever the shape of FDA’s final guidance documents for regulating LDTs, Rheonix believes that its Universal CARD cartridge and Encompass Optimum workstation will remain valuable tools in both research and clinical laboratory settings.
With such a tool in hand, a mutually beneficial partnership of FDA, CMS, IVD manufacturers, and clinical laboratories can encourage the development and effective utilization of LDTs to meet the diverse and growing needs of patients, physicians, and payors.
Richard A. Montagna, PhD, FACB, is senior vice president for scientific and clinical affairs at Rheonix Inc. For further information contact CLP chief editor Steve Halasey at [email protected].
REFERENCES
- Draft Guidance for Industry, Food and Drug Administration Staff, and Clinical Laboratories: Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs) [Framework Guidance]. Silver Spring, Md: Office of In Vitro Diagnostics and Radiological Health, Center for Devices and Radiological Health, FDA, 2014. Available at: www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm416685.pdf. Accessed September 7, 2016.
- Muldrew KL. Molecular diagnostics of infectious diseases. Curr Opin Pediatr. 2009;21(1):102–111; doi: 10.1097/mop.0b013e328320d87e.
- Macgregor PF, Squire JA. Application of microarrays to the analysis of gene expression in cancer. Clin Chem. 2002;48(8):1170–1177.
- Gage BF, Eby C, Johnson JA, et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther. 2008;84(3):326–331; doi: 10.1038/clpt.2008.10.
- Ochiai T, Nishimura K, Watanabe T, et al. Identification of responders/non-responders to 5-fluorouracil based on individual 50% inhibitory area under the concentration curve of 5-fluorouracil obtained with collagen gel droplet-embedded culture-drug sensitivity test in colorectal cancer. Oncol Lett. 2011;2(2):309–313; doi: 10.3892/ol.2011.251.
- Mathew C. Science, medicine, and the future: postgenomic technologies: hunting the genes for common disorders. BMJ. 2001;322(7293):1031–1034.
- Medicare will use private payor prices to set payment rates for clinical diagnostic laboratory tests starting in 2018 [press release online]. Baltimore: Centers for Medicare & Medicaid Services, 2016. Available at: www.cms.gov/newsroom/mediareleasedatabase/press-releases/2016-press-releases-items/2016-06-17.html. Accessed September 13, 2016.
- Draft Guidance for Industry, Food and Drug Administration Staff, and Clinical Laboratories: FDA Notification and Medical Device Reporting for Laboratory Developed Tests (LDTs) [Notification Guidance]. Silver Spring, Md: Office of In Vitro Diagnostics and Radiological Health, Center for Devices and Radiological Health, FDA, 2014. Available at: www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm416684.pdf. Accessed September 13, 2016.



{kind=link}