
Whole-genome copy-number assays advance solid tumor management
By Kabir Chaturvedi

Kabir Chaturvedi, Affymetrix
Expanding knowledge of the human genome has dramatically accelerated the approach of medical professionals to cancer diagnosis and treatment. Vast tracts of human DNA sequences have been implicated in causing cancer, driving its progression, and conferring both response and resistance to therapy. Such discoveries have only recently become possible as a result of advances in whole-genome technology, such as copy-number assays compatible with formalin-fixed, paraffin-embedded (FFPE) specimen preservation.
Concurrently, these technologies have also benefited from improvements in workflow, ease of use, and the interpretability of results, thereby improving the pace at which new discoveries in the lab are transmitted to patients. Until a decade and a half ago, understanding a patient’s genetic makeup would have required the involvement of scores of scientists, and would have taken months to complete. Today, it is possible for a single technician to paint a whole-genome portrait of a patient’s total molecular aberrations within just 48 hours. As a result, the cancer community stands to make significant progress against cancers driven by copy-number aberrations through better diagnosis, prognosis, and therapy selection.
ADVANCING PRACTICES
Historically, a barrier to optimal analysis of solid tumor samples was the standard process whereby a tumor biopsy sample is placed in FFPE blocks for transportation and subsequent storage. This practice presented challenges because small and precious samples can become degraded during FFPE preservation, and consequently generate low-input DNA.

The OncoScan FFPE assay kit by Affymetrix Inc
Additionally, solid tumor samples often are low in purity because they are contaminated with normal tissue, and the tumor is heterogeneous (only a fraction of cells within the tumor may contain the aberration). To detect such copy-number and somatic mutations that are located in a small fraction of cells in the sample, highly sensitive technology is required. The accepted approach for most solid tumor characterization has involved immunohistochemistry and fluorescent in situ hybridization (FISH) staining.
In the past few years, there has been significant attention paid to the emerging options of whole-genome and whole-exome next-generation sequencing (NGS) as methods for detecting mutations in solid tumor samples. However, these techniques require significant overhead in equipment, personnel training, data analysis, and turnaround time, making them impractical for most routine clinical and research applications. NGS systems are expensive (several hundreds of thousands of dollars per instrument), and running them requires sophisticated computing resources—both human and machine. In the best of circumstances, completing the assembly of a whole human genome can take several weeks, requiring the use of scarce central processing unit time. It is not uncommon for the process to encounter delays lasting several months—far too long for a cancer patient who is waiting for a diagnosis or to be placed on an efficacious therapy regimen.
CLASSIFYING CANCERS
Genetic sequencing has come to be used routinely for discovering somatic mutations that contribute to cancer. But recent research findings have uncovered a second class of tumors that is characterized by recurrent copy-number changes. This discovery has opened up the possibility for new diagnostic workflows, new approaches to patient stratification, and new targets for cancer therapy. Among diagnostics researchers, the ultimate goal is to discover greater numbers of high-quality biomarkers for diagnosis, prognosis, and prediction of response to therapy.
In 2006, the National Institutes of Health launched the Cancer Genome Atlas (TCGA), a pilot project to support research and create a comprehensive atlas of genomic changes involved in cancer. A national team of researchers collaborated on the project with the aim of making the data available to scientists around the world. One of the significant findings of this work, communicated in a recent paper in Nature Genetics, is that solid tumors can be divided into two categories: M-class tumors, characterized by somatic mutations; and C-class tumors, characterized by multiple recurrent copy-number changes (gains or losses) but not somatic mutations.1
Importantly, this research also revealed that cancer-related genetic alterations are shared among C-class tumors, independent of tumor tissue type. It turns out that C-class tumors include almost all ovarian and breast cancers, and a large fraction of lung and head and neck squamous cell carcinomas and serous endometroid tumors.

The Affymetrix OncoScan FFPE assay is read on the company’s GeneChip Scanner 3000.
In light of these recent discoveries, it is clear that somatic mutations may tell only a part of a tumor’s story. When testing is limited to uncovering somatic mutations, as it is with most sequencing, important “druggable” target biomarkers may be missed, especially in C-class tumors. Focal amplifications and deletions of critical gene regions are another important source of molecular information, which can be overlaid with expression profiling to give a picture of actionable markers. Focal events can affect either transcription or translation of downstream oncogenes, which may in turn affect progression of disease.
Knowledge of these molecular switches could thus facilitate therapeutic intervention, steering patients toward a more efficacious treatment regimen. For example, patients treated with anti-epidermal growth factor receptor (EGFR) therapies in colorectal cancer ultimately develop resistance and progress in disease state. However, detection of focal amplifications of KRAS, MET, and ERBB gene loci at a whole-genome level can serve as an early warning of molecular resistance to anti-EGFR therapy. The patient can then be placed on an alternative regimen, thereby saving precious time and increasing the likelihood of continued response to therapy. The TCGA paper and other publications list at least 50 actionable or ”druggable” copy-number markers known today, and this list is growing.
Whole-genome analysis is necessary for the discovery of novel biomarkers because recurrent aberrations associated with clinical outcomes may be in any part of the genome. Targeted genomic approaches typically query only 200 to 400 genes and may miss these changes. Furthermore, whole-genome approaches obviate the need for targeted gene lists, which evolve over time as new genes are added and removed based on functional importance. Having whole-genome data is a useful reference that can be mapped to any gene list in the future. This approach saves researchers from having to revalidate their targeted assays every few years. Targeted NGS panels, which interrogate small stretches of the genome and are cheaper than whole-genome sequencing, struggle with sequence-capture bias and, as a result, lack the sensitivity to detect low-level copy-number variants (less than eight copies) that are proven to be actionable markers for disease. This is true even at sequencing depths greater than or equal to 500x coverage.
AN ARRAY-BASED APPROACH
A new technology called the molecular inversion probe (MIP), developed by Affymetrix, Santa Clara, Calif, addresses the challenges discussed above with an array-based multiplex assay (the OncoScan FFPE assay kit) that delivers whole-genome copy number analysis in just 48 hours from as little as 80 ng of DNA.
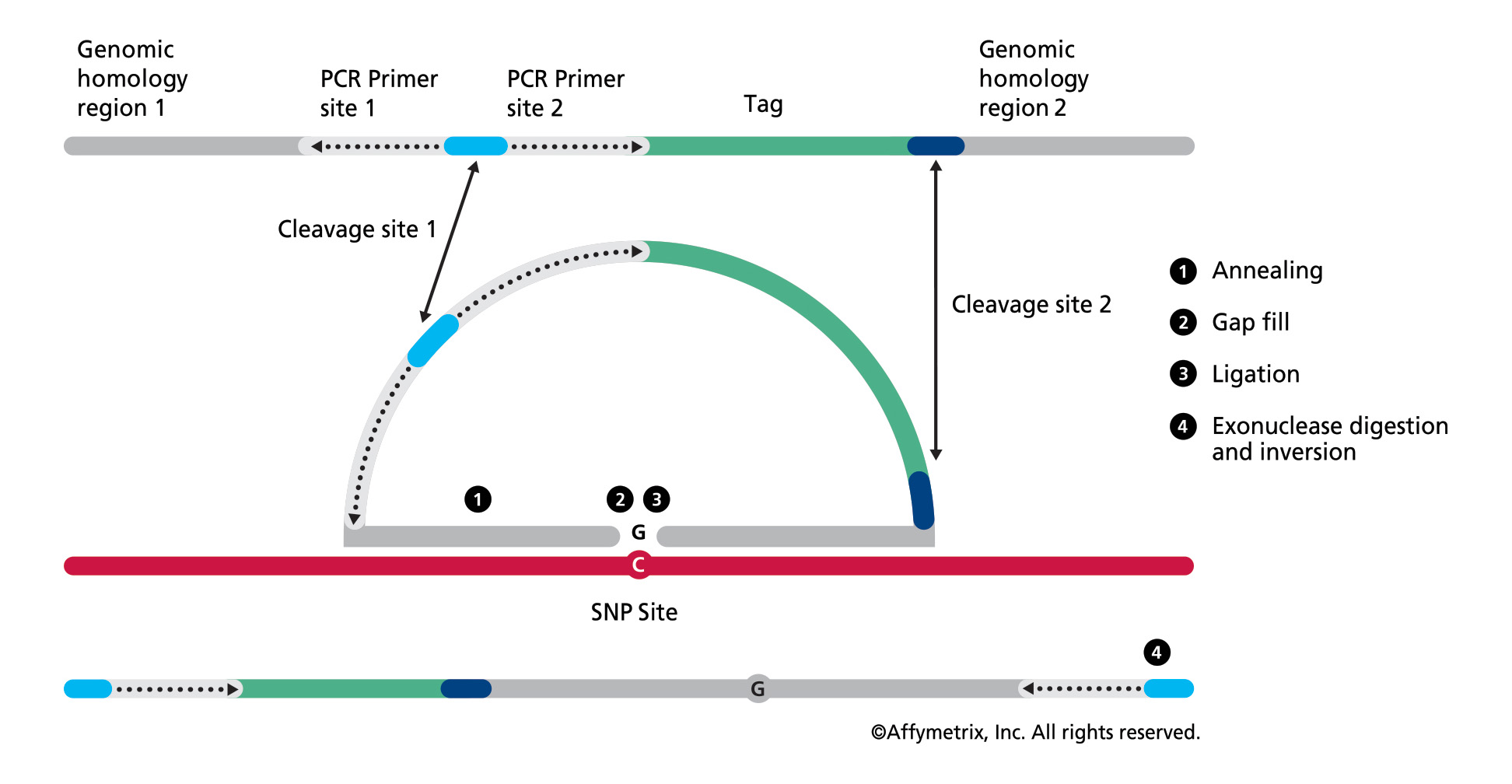
Figure 1. In the OncoScan FFPE assay by Affymetrix, the molecular inversion probe technology interrogates 40 base pairs of genomic DNA, using padlock probes that hybridize to the target of interest before polymerase chain reaction amplification, leading to high assay specificity. Results are available in 48 hours. (Click to enlarge)
Central to the MIP assay technology are padlock probes that hybridize to the DNA target of interest before polymerase chain reaction (PCR) amplification, leading to high assay specificity (see Figure 1). The OncoScan assay provides whole-genome copy-number data with specifically enhanced high resolution for approximately 900 known cancer genes (detecting the addition or deletion of these oncogenes), loss of heterozygosity across the whole genome, and clinically relevant mutation data. Suited for use with highly degraded FFPE solid tumor samples, the assay is enabling advances in discovery and therapy. Some examples of work being done by institutions utilizing this technology include the following:
- ARUP Laboratories, Salt Lake City, was one of the early adopters of the OncoScan FFPE assay kit, and has been using it on multiple solid tumor tissue types, including melanoma, glioma, breast carcinoma, and liposarcoma. ARUP has published on the improved diagnostic yield in melanoma from using the OncoScan FFPE assay kit, finding the new technology to be a more multiplexed, higher resolution, and higher specificity alternative to targeted FISH testing.2 This is because malignant melanoma is characterized by large structural changes that are scattered across the whole genome and cannot be detected using targeted approaches. ARUP is using the OncoScan FFPE assay kit as a complementary test to NGS methods, which are used for somatic mutation detection. Taking the information from these complementary sources together, researchers can identify markers of disease diagnosis and prognosis, and recommend therapeutics that are better tailored for the patient’s molecular profile.
- The Institute of Neuropathology, Bonn, Germany, was also an early user of the array technology and recognized its potential in the clinical setting for the detection of specific copy-number gains and losses, and loss of heterozygosity in brain tumors, as markers for patient stratification and response to treatment. Glioblastoma is a disease in which a whole-genome approach is essential because many of the clinically relevant aberrations are large structural changes scattered across the whole genome. This organization has used the OncoScan assay to help in molecular staging of patient brain tumor samples as well as stratifying patients into different molecular subtypes for treatment.
- Researchers at Washington University, St Louis, used the OncoScan FFPE assay kit to develop a whole-genome profile of rhabdoid and clear-cell carcinomas, which are variants of renal cancer that are very difficult to distinguish under the microscope using immunohistochemical staining. This is also a tumor type where a whole-genome approach is needed because several of the clinically relevant alterations are large structural changes spread throughout the genome.
In addition to delivering key copy-number information, the array-based approach provides significant workflow advantages compared to NGS. It is faster and less expensive, requiring little training and low overhead. Delivering results in just 48 hours, the MIP technology is becoming a tool of choice for the analysis of FFPE cancer tumor samples. These new tools are enabling rapid analysis and answers about which genes are actionable and are contributing to therapeutic drug decisions.
CONCLUSION
Whole-genome copy-number analysis is shifting the paradigm of cancer patient diagnosis, prognosis, and therapy selection. Novel biomarkers are being analyzed that could not previously have been detected using a targeted approach. When combined with data about somatic mutations, data about copy-number alterations can offer a more comprehensive snapshot of the molecular etiology of cancer. Technology selection should be based on compatibility with key clinical laboratory procedures and objectives, including compatibility with FFPE fixation, ease of use, and rapid turnaround time.
Kabir Chaturvedi has strategic marketing responsibilities for oncology clinical research products at Affymetrix Inc, Santa Clara, Calif. For more information, contact CLP chief editor Steve Halasey via [email protected].
REFERENCES
1. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45(10):1127–1133.
2. Rowe LR, Chandler W, Jahromi M, Schiffman JD, South ST. Copy number, loss of heterozygosity and amplification detection in formalin-fixed paraffin-embedded melanocytic lesions using molecular inversion probes. Cancer Genetics. 2011;204(8):473; doi: http://dx.doi.org/10.1016/j.cancergen.2011.08.010.


{kind=link}