
Evaluating risk is the backbone of individualized quality control planning
By Rose Mary Casados, BSMT(ASCP)

In the delivery of quality patient care, laboratory medicine prides itself on performing extensive, effective, and documented quality control (QC), giving healthcare professionals confidence that test results obtained on patient specimens are consistently accurate and reliable. Diagnostic quality control has evolved as laboratory instrumentation and testing technologies have advanced.
The Clinical Laboratory Improvement Amendments of 1988 (CLIA) are the federal regulations that govern all US laboratory testing used for the “diagnosis, prevention, or treatment of any disease or impairment of the health of human beings.”1 In 1992, when the initial amendments became effective, the minimum requirement for QC established for laboratory diagnostics was to perform two levels of control materials each day of patient testing. However, in 2003, when final CLIA regulations were published, a set of “Interpretive Guidelines for Laboratories” (Appendix C) allowed for an alternative to the performance of daily external quality control, so long as “equivalent quality testing” is conducted.2
The Appendix C guidelines permitted the Centers for Medicare and Medicaid Services (CMS) to modify its interpretation of the CLIA regulations without making revisions to the actual law, effectively paving the way for new QC ideas, methodologies, and technologies to be considered. In turn, CMS implemented the policy of accepting laboratories’ equivalent quality control (EQC) procedures, which enabled laboratories to decrease their frequency of performing external controls once a successful standardized qualifying study had been performed and documented.
With continued focus on improving quality measures, CMS subsequently challenged the laboratory community to develop a new QC guideline by convening a meeting focusing on “QC for the Future.” In attendance were accrediting organizations (ie, COLA, the College of American Pathologists, and the Joint Commission), government agencies, and industry representatives. The result—a laboratory quality control guideline based on risk management—was used to develop a new alternative QC policy, called Individualized Quality Control Plan (IQCP), which is scheduled to replace EQC on January 1, 2016. It is anticipated that laboratories that formerly used EQC will implement IQCP.
Prior to discussing the elements of an IQCP, it is important to understand that IQCP is quality control based on risk management. By definition, “risk” is a measure of the severity of the impact of a potential error, multiplied by the probability of how likely it is that the error will occur and the ability to detect the error if it should occur. Correspondingly, “risk management” is the sequential process of risk identification, risk assessment, and risk mitigation. Simply stated, risk management includes:
- Risk identification: identifying potential errors;
- Risk assessment: evaluating errors to determine their impact on patient test results; and
- Risk mitigation: controlling errors in such a way that residual risk is manageable.
This article will review the first two phases of this process—risk identification and risk assessment—with an understanding that subsequent to identifying the risk and assessing the severity of the risk, risk mitigation is the ultimate goal.
RISK IDENTIFICATION
Risk identification is the first and perhaps the most important step in the risk management process. Correctly identifying potential sources of error for a particular test reveals valuable information for developing an individualized quality control plan. Identifying the potential risks embedded in laboratory testing processes allows for the implementation of an overarching quality control plan that effectively mitigates errors not addressed by the test system’s internal checks and external quality control.
Medical decisions and effective treatment planning are dependent on laboratory results. Therefore, it is important to understand that in performing an effective risk assessment, “one size does not fit all!” Every diagnostic test carries varied risks and may require varied control measures. Accurate risk identification is essential when developing and implementing a comprehensive and effective individualized quality control plan.
There are several tools available to help laboratorians perform risk identification. The most effective tool will vary from laboratory to laboratory. Therefore, it is important that all options are considered, and that the most effective tool is selected that will positively impact the uniqueness of each laboratory. The following are examples:
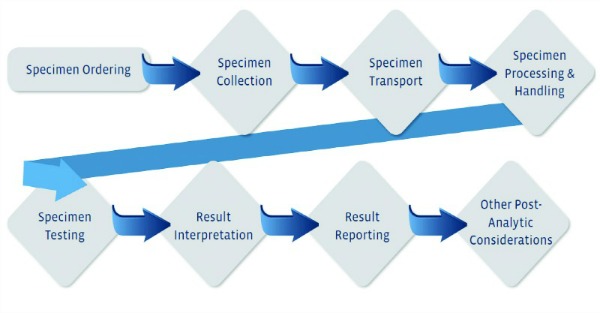
- Process mapping: A process map is a graphical representation of all the steps in a testing process. This tool is used to analyze a particular testing process by breaking it down into small steps from start to finish (see Figure 1).
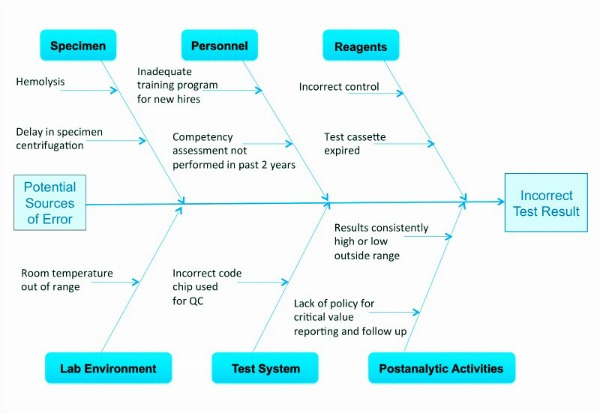
- Fishbone diagram: A fishbone diagram outlines the cause and effect of a testing process. It may be considered to be a graphical representation of all the major elements in a process. This diagram can help labs identify and organize potential errors in a test system (see Figure 2).
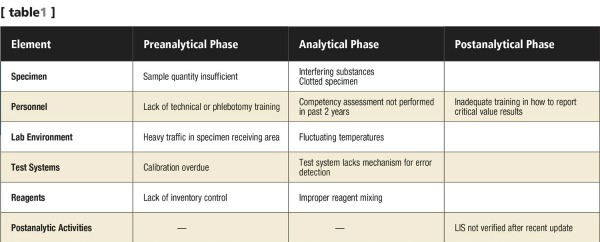
- Risk identification table: A risk identification table is a simple table that lists all the errors identified in the different testing phases for a specific test (see Table 1).
Regardless of the tool selected, it is important to consider all phases of sample testing: the preanalytic, analytic, and postanalytic phases. The key objectives in implementing an effective risk management process and, hence, an IQCP, are identifying the potential errors in all phases of testing, and ensuring optimal mitigation by putting in place effective and documented processes.
Results obtained in performing the risk assessment will be used to develop an IQCP. This plan will serve to describe QC practices, resources, and procedures that will be used to monitor and ensure continued quality laboratory testing. As with all laboratory procedures, the laboratory director remains ultimately responsible for the proper development and implementation of each IQCP. The laboratory director is required to approve and sign each IQCP before it is implemented.
RISK ASSESSMENT
Although the goal for all healthcare professionals is to optimize the delivery of quality patient care, error rates are evident and distributed among the three phases of testing: preanalytic, analytic, and postanalytic.
For example, studies have shown that 46% to 68.2% of lab errors occur during the preanalytic phase. Such errors may include, but are not limited to, inappropriate test requests, order entry errors, misidentification of patients, utilization of improper containers, inadequate sample collection and transport procedures, inadequate sample/anticoagulant volume ratio, insufficient sample volume, sorting and routing errors, and labeling errors.
By contrast, roughly 7% to 13% of lab errors occur during the analytic phase. Such errors may include, but are not limited to, equipment malfunction, sample mix interference, undetected failure in quality control, and incorrect testing procedure.
And finally, 18.5% to 47% of errors occur in the postanalytic phase. Such errors may include, but are not limited to, failure in reporting, erroneous validation of analytical data, and improper data entry and reporting.3
From these examples, one can conclude that greater emphasis has traditionally been placed on risk identification and risk mitigation for the analytic phase of testing versus the preanalytic and postanalytic phases. However, the high error rates apparent in the preanalytic and postanalytic phases demonstrate precisely the need for risk management in all phases of testing. Risk management is an all-inclusive quality management process that guides the laboratorian to evaluate potential risks in all phases of testing, which is the essence of IQCP.
When performing a risk management process and, hence, evaluating the entire testing process, it is important to consider the following key areas within the five components of the diagnostic process that affect the quality patient test results: environment, testing personnel, specimen, testing process, and reagents.
In doing so, how the test is performed, how often relevant errors or undesirable conditions occur, the potential impact of those errors, what control activities are in place to detect or prevent those errors, how the test is used by the physician, patient population, and volume of testing can all be considered in developing the risk assessment. The following sections offer some key examples of risk assessment considerations in each of the five areas.
Environment. Areas of importance that require focus include the following:
- Where is testing performed, and what other activities occur nearby?
- Are room temperature and humidity stable?
- Are testing areas level, and draft- and vibration-free?
- Does altitude affect the testing?
- Have lighting, electricity, water quality, and other utilities been considered?
Testing Personnel. Individuals performing testing must be evaluated to ensure that their training and competency assessment records can be used to validate the qualifications of testing personnel to accurately perform testing. Key questions to be addressed in this area include the following:
- Do testing personnel have laboratory medicine education and experience?
- Have testing personnel been adequately trained to perform the test?
- Have competency assessments been performed on testing personnel?
Although testing personnel may have been trained to perform specific types of testing, it cannot be assumed that all testing personnel maintain the superior level of performance they initially demonstrated. It is therefore essential that competency assessments be performed on a continuing basis.
Specimen. Ensuring the collection of the right specimen from the right patient is essential.
- Review specimen collection, handling, and storage procedures.
- Review all instructions that are provided to patients regarding preparation for the self-collection of specimens.
It is important to ensure that all specimens are handled and stored appropriately, and are suitable for testing.
Testing Process. It is important to gather information regarding the measuring system, the instrument, or other test device. Varied types of information can contribute to risk assessment in this area.
- For commercialized instruments, tests, and test kits, a great deal of such information is supplied by the manufacturer through a package insert or operator’s manual.
- A test system may have safeguards such as lockout functions and error codes that detect and prevent errors.
- Laboratory historical data can be considered. Maintenance logs, QA and QC records, verification of performance specifications, and calibration verification records are a few examples of documentation to review.
In addition, an assessment of risk should consider the utilization of test results by the ordering physician when developing a treatment plan for the patient. Some test results represent only a portion of the information that contributes to clinical decisions. Others are used immediately, as the sole decision-making criterion. In the latter situation, an inaccurate result has a much greater potential to cause harm, making an understanding of the clinical use of test results an important element for determining the quality control measures a lab needs to perform.
Reagents. Test reagents can be compromised during shipment, handling, storage, and processing. In addition, consideration should be given to the quality and stability of reagents used directly as part of the QC process:
- Calibrators
- Controls
It is important that package inserts and storage records are gathered and reviewed to assess these potential risks.
CONCLUSION
Although risk management may appear menacing, laboratorians will soon realize that risk management simply encompasses management processes focused on the delivery of quality laboratory medicine, which have historically been performed on a daily basis. Having performed risk identification and risk assessment sequentially, laboratorians can move on to framing the ultimate outcome of the process—risk mitigation—which will result in the successful completion of an IQCP.
Developing a risk management process does not ensure the complete elimination of risk. However, implementing a thorough risk management process that includes detailed risk identification and assessment will contribute to the reduction of risk and result in the continued delivery of quality patient care.
Rose Mary Casados, BSMT(ASCP), is president of COLA Resources Inc (CRI), an educational and training subsidiary of laboratory accreditor COLA. CRI offers educational resources to help labs transition to the new IQCP environment, including the IQCP E-Optimizer, a software tool that provides laboratories with a guide on how to perform risk assessment and develop an IQCP; and the CRI Implementation Guide, an all-inclusive manual that assists laboratories in implementing an IQCP specific to their needs. For further information, contact CLP chief editor Steve Halasey via [email protected].
REFERENCES
1. Clinical Laboratory Improvement Amendments, Subpart A, Section 493.1 Basis and scope. Available at: http://www.gpo.gov/fdsys/pkg/CFR-2011-title42-vol5/pdf/CFR-2011-title42-vol5-part493.pdf. Accessed January 25, 2015.
2. Centers for Medicare and Medicaid Services. Clinical Laboratory Improvement Amendments, Interpretive Guidelines for Laboratories. Available at: http://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/Interpretive_Guidelines_for_Laboratories.html. Accessed January 25, 2015.
3. Hammerling JA. A review of medical errors in laboratory diagnostics and where we are today. LabMed. 2012;43(2):41–44. Available at: http://labmed.ascpjournals.org/content/43/2/41/T1.expansion.ht; doi: 10.1309/lm6er9wjr1ihqauy. Accessed January 26, 2015.



{kind=link}