
Patients with rare genetic diseases can face a difficult path to a proper diagnosis. Because there is a lower awareness of such diseases, and because they have symptoms that may initially be confused with more common diseases, diagnosis can take several years, with patients often consulting with multiple physicians. Access to comprehensive diagnostic testing can also be a barrier to diagnosis (Figure 1).
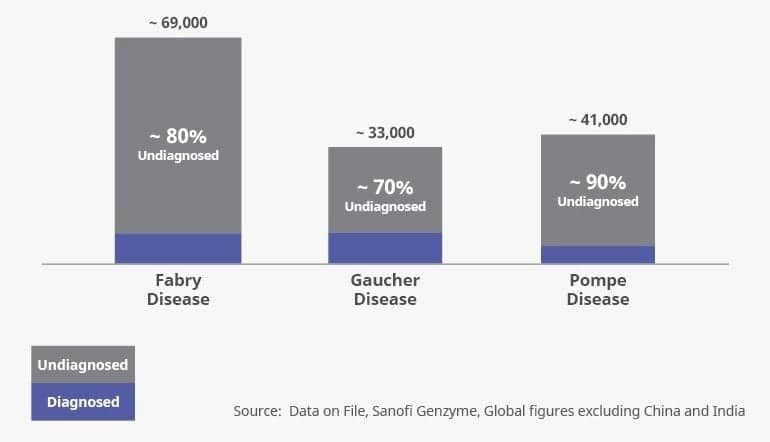
Figure 1. The rare disease diagnosis gap.
The Lantern Project aims to reduce such barriers by providing a no-cost testing program for patients in the United States who suffer from specific types of lysosomal storage disorders. Physicians will be able to use the Lantern Project to arrange for screening of certain suspected disorders as well as confirmatory DNA testing and phlebotomy services for their patients.
Launched by Sanofi Genzyme, Cambridge, Mass, in partnership with PerkinElmer Genomics, Waltham, Mass, the Lantern Project will assist patients whom physicians suspect may be suffering from Fabry disease, Gaucher disease, mucopolysaccharidosis type I (MPS I), Niemann-Pick disease types A and B (acid sphingomyelinase deficiency; ASMD), or Pompe disease. In addition, the project will offer the option of an enzyme panel for seven mucopolysaccharidoses, and a 105-gene panel for limb-girdle muscular dystrophies (LGMD) and other myopathies.
“The Lantern Project is another reflection of Sanofi Genzyme’s continuing commitment to the rare disease communities we serve,” says Sarah Gonzalez, head of medical diagnostics at Sanofi Genzyme, the specialty-care global business unit of Sanofi.“ While we have seen many significant advances in research over the past 30-plus years, there are still tremendous challenges in helping patients get a diagnosis for many rare diseases.”
Barriers to Early Diagnosis
Lysosomal storage disorders comprise a group of more than 40 genetic diseases. People living with such disorders experience a deficiency or malfunction in an enzyme needed to remove unnecessary waste material from cells in the body. The waste molecules accumulate in cells, disrupting cell function and leading to progressive organ damage.
Lysosomal storage disorders are frequently characterized by a range of symptoms that can present at any time from early childhood to late in adulthood. Both disease progression and symptom severity can vary widely. The fact that such diseases are often rare and have symptoms associated with other more common diseases increases the risk of misdiagnosis, lengthy diagnostic delays, and missed cases.1 In addition to putting patients at risk of further disease progression, delay in diagnosis can lead to feelings of anxiety, frustration, and isolation.
The Lantern Project
For more than a decade, Sanofi Genzyme has supported a range of programs to help patients have broader access to genetic testing services for specific types of lysosomal storage disorders.
The Lantern Project is specifically designed to overcome the barriers that patients experience in accessing comprehensive testing services for those diseases, including additional diagnostic tests that may be needed when the results of genetic testing are suggestive but inconclusive (Figure 2).
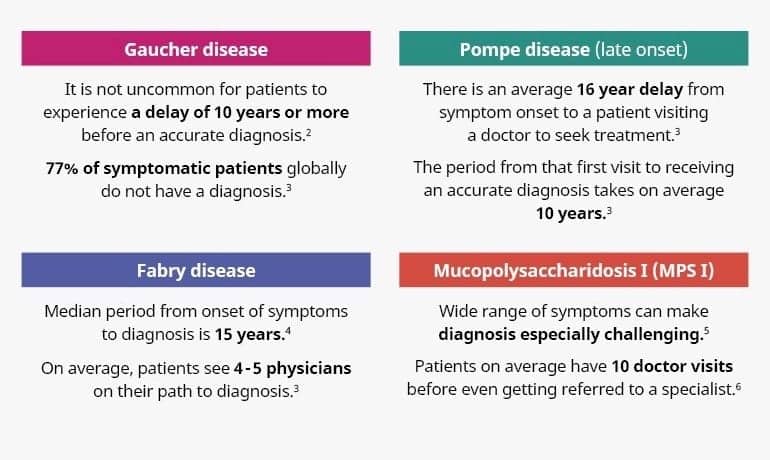
Figure 2. Challenges in rare disease diagnosis.
One of the highlights of the program is the LGMD panel, which is able to sequence 105 genes known to be associated with LGMDs and other myopathies, a heterogeneous group of muscular weakness disorders that vary in severity and age of onset. Using next-generation sequencing technology that allows for rapid sequencing of multiple genes simultaneously, patients can be tested for multiple LGMD disorders at once in addition to other diseases that may cause similar symptoms, including Pompe disease and spinal muscular atrophy.
Genetic testing can play an important role in confirming a diagnosis for patients living with many lysosomal storage disorders. Early diagnosis can also help patients advance to more timely initiation of options in disease management.
The Lantern Project is not intended to and should not interfere in any way with a healthcare professional’s or patient’s independent judgment and freedom of choice in the treatment options for these diseases. Healthcare professionals and patients should always consider the full range of treatment options and select those most appropriate for the individual patient.
“The Lantern Project will help more patients access testing while also raising broader awareness of the critical need for more diagnostic services and support for all people affected by rare diseases,” says Gonzalez.
For more information, visit the Lantern Project.
References
- Kingma SD, Bodamer OA, Wijburg FA. Epidemiology and diagnosis of lysosomal storage disorders: challenges of screening. Best Pract Res Clin Endocrinol Metab. 2015;29(2):145–157; doi: 10.1016/j.beem.2014.08.004.
- Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am. J. Hematol. 2007;82(8):697–701.
- Sanofi Genzyme market research and internal analysis; data on file.
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30; doi: 10.1186/1750-1172-5-30.
- MPS I (Hurler, Hurler-Scheie, Scheie syndrome) [online]. Durham, NC: MPS Society, 2017. Available at: http://mpssociety.org/mps/mps-i. Accessed July 1, 2018.
- Bruni S, Lavery C, Broomfield A. The diagnostic journey of patients with mucopolysaccharidosis I: A real-world survey of patient and physician experiences. Mol Genet Metab Rep. 2016;8:67–73; doi: 10.1016/j.ymgmr.2016.07.006.
Featured image: Illustration of alpha-galactosidase (agalsidase) enzyme, the cause of Fabry’s disease. Image courtesy Dreamstime.




{kind=link}
It is great blog post. I am Always read your blog. Helpful and Informative blog. Thanks for sharing these information with us.