
 Reimbursement looms as major challenge
Reimbursement looms as major challenge
BY: GARY TUFEL
Personalized medicine is the process of subsetting patient populations, in some cases down to the individual, using diagnostics that identify genetic and other proclivities for or against the use of specific therapies to treat specific conditions in specific patients, notes Alan Wright, MD, chief medical officer, Roche Diagnostics, Indianapolis.
It has enormous implications, from enabling the identification and treatment of more common diseases, to diagnosing and treating cancers due to particular genetic abnormalities or mutations, to rare genetic diseases found only in particular ethnic groups. Wright says personalized medicine improves patient response rates and provides a better understanding of which individuals will respond to certain treatments, allowing for more targeted therapies.
REALISTIC PRICING FOR DIAGNOSTICS
Yet despite the critical role diagnostic tests play in personalized medicine, they are significantly undervalued, Wright says, and the disparity that exists between current investment models and reimbursement policies for diagnostics is not tenable long-term.
“We hear about the cost of technology declining and diagnostic tests becoming more affordable, but that is only half the story,” Wright says. “In the world of personalized medicine, diagnostics should be valued on the basis of the R&D investment that’s required to develop a clinically validated test.”
Wright cites the example of the pharmaceutical industry, where drugs are valued according to the investment in research, which according to a February 2012 Forbes article can cost more than $1 billion per drug and take more than a decade to develop. The cost of development is amortized over the patent life span of the drug in order to arrive at a price. Similarly, Wright says, the process of developing an FDA-approved diagnostic test for clinical use costs tens of millions of dollars and spans several years. But the pricing model is decidedly different.
 |
“Laboratory testing and clinical validation are part of the ‘personalized’ in personalized medicine. But it follows that as science helps define the uniqueness of each person’s condition, disease, or risk profile, the potential volume for each specific test in the population will be less.”
—Alan Wright, MD, chief medical officer, Roche Diagnostics
|
Through techniques such as “code stacking,” “crosswalking,” and “gap filling,” pricing for diagnostics is essentially determined by what it costs to perform the test in the lab. These prices are fixed by the federal government and by payors. “While manufacturers could theoretically charge more for reagents to recover the costs of research,” Wright notes, “that strategy would quickly squeeze the laboratory because reimbursement levels are fixed.” For tests that have an intended use across a broad population base, volume can help a manufacturer recover the costs. But as personalized medicine narrows the focus of specific diagnostics and therapies to smaller subsets of the population, the potential market and potential return on investment for individual tests are shrinking along with it. For a robust diagnostics industry to remain viable, Wright says, the available commercial opportunity must be greater than the cost of developing a test and confirming its clinical utility.
“Laboratory testing and clinical validation are part of the ‘personalized’ in personalized medicine,” he says. “But it follows that as science helps define the uniqueness of each person’s condition, disease, or risk profile, the potential volume for each specific test in the population will be less.” When Roche introduced an FDA-approved BRAF mutation test in 2011 as a companion diagnostic to identify patients eligible for a specific therapy for metastatic melanoma, he says, the development cost tens of millions of dollars. But the typical reimbursement level to the lab is only $130 or less, and the population of patients who qualify for the test each year is relatively small. “To maintain investment from the private sector in diagnostics, we need a more attractive path to market,” Wright says.
That private investment currently makes up more than 50% of the funding for life science research, which makes a significant contribution to medical innovation and helps demonstrate the clinical utility of FDA-approved tests. Wright says a lack of funding for clinical trials would likely lead to less robust studies and a weaker correlation between test results and clinical outcomes.
CHALLENGES FOR ALTERNATE PRICING STRATEGIES
Of course, labs can develop their own tests without clinical validation studies. But that can lead to issues with a lack of standardization in results and treatment. “Labs using different techniques will get different results, and patient treatment will vary based on the quality of the lab test,” Wright says. “IVD tests are required to have more rigorous clinical validation, and the studies must demonstrate that the test is reliable and works the same way every time.” These FDA-approved studies, he says, must link the particular test to a specific clinical outcome, such as improvement from therapy or reduced risk of disease or death.
As an example, Wright notes that Roche’s ATHENA cervical cancer screening trial, which included more than 40,000 women, was an expensive undertaking but yielded such robust and compelling data related to high-risk HPV testing that the results contributed to the development of new cervical cancer screening guidelines by the American College of Obstetricians and Gynecologists and several other influential groups. But as eligible populations shrink with the development of highly targeted diagnostics (Wright notes that there are more than 90 types of leukemia to test for), it’s no longer commercially viable for manufacturers to do extensive randomized-control trials for all of their tests.
This conundrum of personalized medicine is an issue that the industry, including such groups as the Centers for Medicare and Medicaid Services (CMS) and the American Society of Clinical Oncology, is grappling with, he notes. For meaningful change to occur, policy reform will be required, and thus discussions are under way between industry, academia, and government regarding ways to decrease the cost of clinical development. “Though there are no conclusions and the discussion is in its formative stages, everyone agrees that it is a good thing to have a vibrant, independent diagnostics industry,” Wright says. “The discussions have focused on alternative study designs and tiered risk structures for FDA oversight in order to decrease the cost of trials while maintaining quality. While that is an important conversation, policies should also be developed that permit innovators to recover their development costs. That way, they can continue to fund the kind of groundbreaking life science research that led to the evolution of personalized medicine in the first place.”
The FDA recently announced its approval of Tarceva (erlotinib) for its use in first-line treatment of non-small cell lung cancer (NSCLC) along with a companion diagnostic, the cobas EGFR Mutation Test, from Roche Molecular Diagnostics, Pleasanton, Calif. This is the first FDA-approved companion diagnostic that detects epidermal growth factor receptor (EGFR) gene mutations, which are present in about 10% of NSCLC.
 |
The Luminex collaboration with Merck, unlike other Alzheimer’s clinical trials that have failed because they came too late in the disease process, is about being early in the process—that is, it addresses mild cognitive impairment. “It’s a perfect example of companion diagnostics, screening out those who won’t benefit from it from those who will.”
|
ONE COMPANY’S EVOLVING APPROACH
Historically, Luminex Corp, Austin, Tex, internally developed diagnostic products that focused on infectious diseases, but more recently, personalized medicine has come to the forefront, says Tim Dehne, vice president, global marketing.
The company’s flexible, bead-based technology, xMAP, can detect proteins and nucleic acids, he says. Luminex also partners with companies in both the clinical diagnostic and life science research fields. For example, the new Bio-Plex Pro™ Human Cancer Biomarker Panel 1 from Bio-Rad Laboratories, Hercules, Calif, is a magnetic bead-based assay based on Luminex’s xMAP® technology. This research assay panel allows labs to multiplex assays for 16 biomarker proteins known to play roles in key processes such as angiogenesis, metastasis, cell proliferation, cell adhesion/migration, apoptosis, and inflammation. The panel includes eight new targets, such as sEGFR, sVEGFR-1, and osteopontin, which are associated with cancer progression.
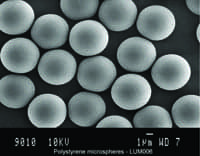 |
| Image of microspheres from Luminex that are used in IVD. Luminex microspheres are used in the company’s most recently FDA-cleared xTAG Gastrointestinal Pathogen Panel (GPP) assay. |
Luminex is also partnering with Merck, Whitehouse Station, NJ, on a companion diagnostic aimed at identifying likely early-onset Alzheimer’s disease patients who could be candidates for an experimental drug that Merck is developing. The two companies recently signed a collaboration and license agreement to develop a companion diagnostic device that will be evaluated to help screen patients for recruitment into Merck’s clinical development program for MK-8931, a novel oral beta amyloid precursor protein site cleaving enzyme (BACE) inhibitor and Merck’s lead investigational candidate for Alzheimer’s disease. Such drug/diagnostic combinations must be developed in tandem to show the FDA they’re efficacious, Dehne notes.
This collaboration, unlike other Alzheimer’s clinical trials that have failed because they came too late in the disease process, is about being early in the process—that is, it addresses mild cognitive impairment, says Jeremy Bridge-Cook, senior vice president, research and development, Luminex. “It’s a perfect example of companion diagnostics, screening out those who won’t benefit from it from those who will,” he says. A recent economic study showed Alzheimer’s is the number one existing disease, even more prevalent than heart disease, with enormous amounts of money being spent to address its effects, Dehne says.
Luminex is also involved in developing CYP-P450 genetic tests for liver enzymes, which may affect the metabolism and clearance of many drugs on the market today—for many of which one P450 gene can play a significant role in whether a drug is efficacious and safe, Bridge-Cook says. Cardiologists are interested in this area, Dehne adds, because some patients do not metabolize Plavix, and that information is necessary to know before heart surgery is performed. It’s important to know not only if a drug is safe, but whether it’s simply ineffective, he notes.
Despite all of the benefits, personalized medicine continues to present challenges to the clinical lab. One is the decision whether or not to implement such tests. And since many of the tests are not FDA-cleared, labs—not the FDA—must take responsibility for them. Bridge-Cook says these tests are largely the province of such large medical entities as Johns Hopkins Medicine, Baltimore, which, in conjunction with pharmaceutical companies, has a degree of resources and professional expertise to lead the personalized medicine charge. Many labs may not feel they have the necessary expertise, he says.
 |
|
Luminex’s portfolio of instruments that use xMAP Technology for clinical and research applications: MAGPIX®, Luminex® 100/200™, and FLEXMAP 3D®. |
Reimbursement playing catch-up
Reimbursement has fallen farther behind, and it’s a very large issue, Dehne says. There’s a strong trend toward cost-cutting and a demand to prove the effectiveness of personalized medicine. Because this is still a newer area, there isn’t a long history for the government—which sets reimbursement rates—to refer to, he says.
CMS has assigned 114 new molecular diagnostics CPT codes, including personalized medicine. CMS did not set the reimbursement rates, and specifically for personalized medicine tests, directed its contractors to do a clean-slate, ground-up gap-fill analysis of test costs, Dehne says. “When contractors finally posted reimbursement rates, they were very low and not sustainable for a testing lab to run a business,” he says. “Since then, [Medicare carrier] Palmetto [GBA] has raised their rates, but they are still well below the 2012 rates,” he notes.
 |
|
The therascreen® KRAS RGQ PCR Kit from QIAGEN. |
QIAGEN’s Companion DX In Oncology Treatment
The therascreen® KRAS RGQ PCR Kit from QIAGEN, Germantown, Md, is a genetic test designed to detect the presence of seven mutations in the KRAS gene in colorectal cancer cells. In normal tissue, the KRAS protein transmits signals in cells to regulate cell growth and cell death. In colorectal cancer tissue, mutations in the KRAS gene cause an altered form of the K-ras protein and result in abnormal functioning of the protein. This test is FDA-approved to aid physicians in identifying patients with metastatic colorectal cancer for treatment with Erbitux® (cetuximab).
The presence of KRAS mutations in colorectal cancer tissue indicates that the patient may not benefit from treatment with Erbitux. If the test result indicates that the KRAS mutations are absent in the colorectal cancer cells, then the patient may be considered for treatment with Erbitux.
Based on comprehensive analytical studies and clinical trials, the test covers the complete workflow from sample prep to detection technology/Rotor-Gene Q MDx instrument and generation of test reports. This standardized workflow is lab- and operator-independent, so reproducible and objective results are facilitated.
In January, Boehringer (BI), Ingelheim, Germany, and QIAGEN submitted applications to the FDA for the marketing approval of afatinib, for non-small cell lung cancer, and companion diagnostic therascreen EFGR to pick out best responders to the drug. FDA decision for BI and QIAGEN is expected in July as both received priority review.
|
ALK FISH ASSAY DETECTS GENE REARRANGEMENTS “Laboratories should use an ALK FISH assay using dual-labeled break-apart probes for selecting patients for ALK TKI therapy; FISH is considered the gold standard for detecting gene rearrangements,” notes Susan Jewell, PhD, scientific affairs manager, Abbott Molecular, Des Plaines, Ill. |
- MORE ON THIS TOPIC:
View the molecular testing products Tech Guide
See an article on oncology and genetic testing
Read an article on emerging technologies
Gary Tufel is a contributing writer for CLP. For more information, contact Editor Judy O’Rourke, [email protected].



{kind=link}