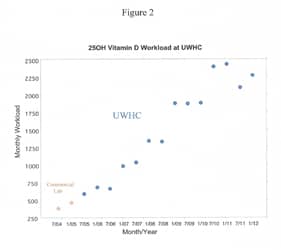
 In the upper left-hand panel, sequencing results for mother, father and daughter are examined, based on multiple reads of a given site in the genome. Each parent is judged to be homozygous—carrying two copies of the same base, (AA). Sequencing of the daughter identifies a heterozygous state at the same site (AC) indicating a de novo mutation, though the new software indicates a very low probability for a mutation at this site, suggesting a false positive result (upper right-hand panel). The lower left and right panels show a failure to identify a heterozygous site in the child, which results in a false negative (with the new method indicating a very high likelihood of mutation at this site).Hidden within the expanse of the human genome, (comprised of some three billion base pairs), mutations are commonplace.
In the upper left-hand panel, sequencing results for mother, father and daughter are examined, based on multiple reads of a given site in the genome. Each parent is judged to be homozygous—carrying two copies of the same base, (AA). Sequencing of the daughter identifies a heterozygous state at the same site (AC) indicating a de novo mutation, though the new software indicates a very low probability for a mutation at this site, suggesting a false positive result (upper right-hand panel). The lower left and right panels show a failure to identify a heterozygous site in the child, which results in a false negative (with the new method indicating a very high likelihood of mutation at this site).Hidden within the expanse of the human genome, (comprised of some three billion base pairs), mutations are commonplace.
While most of them appear to have neutral effect on human health, many others are associated with diseases and disease susceptibility, according to researchers .
Reed Cartwright, a researcher at Arizona State University’s Biodesign Institute, Tempe, Ariz, along with colleagues at ASU, Washington University, St Louis, and the Wellcome Trust Sanger Institute, UK, report on a new software tool known as DeNovoGear, which uses statistical probabilities to help identify mutations and more accurately pinpoint their source and their possible significance for health.
Improvements in the accuracy of mutation identification, and validation could have a profound impact on the diagnosis and treatment of mutation-related diseases, they say.
“These techniques are being considered in two different realms,” Cartwright says. “The first is for pediatric diseases.” Here, a child with an unusual genetic disease may undergo genomic sequencing to see if the mutations observed have been acquired from the parents or are instead, unique to the child. “We can identify these mutations and try to detect which gene may be broken.”
The second application is for cancer research, where tumor tissues are genetically compared with normal tissue. Many now believe that the identification of a specific cancer mutation may eventually permit clinicians to customize a treatment for that tissue type. “We are developing methods to allow researchers to make those types of analyses, using advanced, probabilistic methods,” Cartwright says. “We actually model the whole process.”
The strategy used by researchers, pioneered in part by Donald Conrad, professor in the Department of Genetics at Washington University School of Medicine and corresponding author of the current study, takes advantage of high throughput genetic sequencing to examine whole genome data in search of de novo mutations.
“Our goal is to develop software that will allow researchers and clinicians to estimate a range of mutation types, faster, more accurately, and cheaper,” Cartwright says.
Click here to read the entire story.
[Source: ASU Biodesign Institute]


{kind=link}