When an out-of-control event occurs, laboratories must take immediate and clear action that will identify the root cause, to prevent it from happening again.
By Lorin M. Bachmann and Nico Vandepoele
The most important role of a clinical laboratory is to promptly provide high quality, medically useful laboratory results to healthcare providers. To that end, the laboratory should design and implement an effective quality management system to ensure accurate and reproducible reporting of results. The quality control (QC) program is a component of the laboratory’s overall quality management system. And a routine QC program should be designed to evaluate the analytical performance of the laboratory’s measurement systems to detect potential errors during the analytical phase of testing that may adversely impact patient care.
An out-of-control event occurs when a QC rule evaluation for one or more QC measurements yields unacceptable results. An out-of-control event typically means that the measurement system is not performing within its normal analytical performance specifications. Out-of-control conditions must be detected and investigated to avoid reporting of erroneous laboratory results that may cause patient harm.
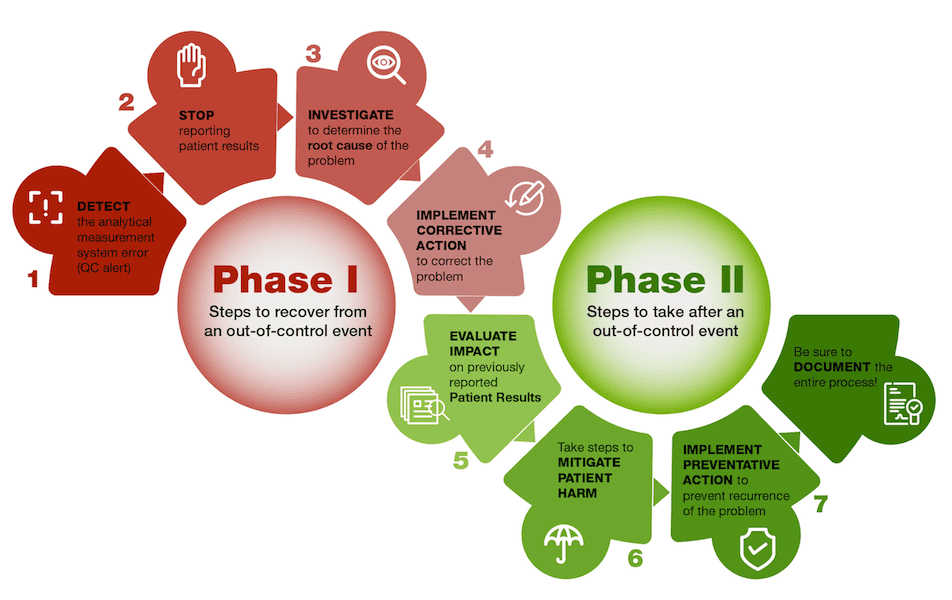
Once an out-of-control event is detected, the challenge is now up to the laboratory to identify the root cause of the event, perform corrective action, mitigate any potential harm to patients and implement preventative action. Following are the step-by-step recommendations for managing out-of-control events, based on recommendations from the Clinical and Laboratories Standards Institute (CLSI) guideline: Statistical Quality Control for Quantitative Measurement Procedures: Principles and Definitions, 4th Edition, CLSI guideline C24.1
Step 1: DETECT the Analytical Measurement System Error
The first step of the response to an analytical measurement system error is to simply identify that it has occurred. An out-of-control event is detected when QC values exceed acceptance limits. An automated QC alert is generated to inform the laboratory of a potential problem, or the laboratory recognizes the result has failed QC acceptance criteria if using manual QC review. Detection of an out-of-control event relies on the establishment of target QC means and standard deviations (SDs) that represent normal performance of the measurement system. The QC target mean should be calculated using a minimum of 10 QC measurements performed over 10 days. This approach ensures that sufficient sources of normal analytical variation, such as variation due to different vials of QC materials, reagent wedge changes and daily maintenance activities are represented in the data used to establish the QC target mean. Calculating the QC target mean using several months’ worth of data is preferable because additional sources of normal variation can be incorporated, such as multiple assay calibrations, reagent lot changes and monthly maintenance activities. The SD is not a function of the QC material, but rather a function of the analytical measurement system performance. Therefore, target SDs should be calculated using several months of historical QC data to incorporate multiple sources of normal variation.
QC rule evaluations can be programmed into QC data management software to automatically alert laboratorians when an out-of-control event is detected. One strategy to establish QC rule evaluations is to base the QC rules on the sigma metric. The sigma metric relates analytical performance to allowable total error (TEa), and its use is described in the CLSI C24 document. If an assay exhibits a large sigma metric, such as six, then the assay has a low error rate. Assays with a low sigma metric, such as three, have a larger error rate. For assays with a low sigma metric, the use of QC multi-rules to detect small shifts or drifts in QC values is desirable.
Step 2: STOP Reporting Patient Results
Once an out-of-control alert is detected, the laboratory should ensure that all patient result reporting is immediately stopped for the affected assay. This could include taking the assay out of service, or even taking the entire instrument out of service, if necessary. If auto verification is performed in the laboratory, auto verification should be turned off so that patient results are not automatically reported into the medical record. Laboratories performing the assay on multiple instruments can re-route patient samples to other measurement systems that have acceptable QC results, while the out-of-control event is investigated.
Step 3: INVESTIGATE to Determine the Root Cause of the Problem
The next step is to perform an investigation to determine the root cause of the out-of-control event. The root cause is the actual reason for the presence of the analytical measurement system error. It is critical to determine the root cause of the out-of-control event, so that the problem can be directly addressed through corrective and preventative actions.
The investigation typically starts with review of QC records and Levey-Jennings charts. Can any obvious user errors be quickly ruled out? For example, was the wrong level of QC material, or the wrong QC product, or the wrong QC lot number analyzed? Was there a sufficient amount of QC material in the sample cup for analysis? Was the QC material left out on the bench at room temperature too long, resulting in degradation of the material?
Once obvious errors are ruled out, the laboratory can conduct more extensive troubleshooting steps. Are there any trends or shifts observed upon examination of the Levey-Jennings charts? Is the out-of-control event related to a recent change in the measurement system such as a recent assay calibration, new reagent lot or maintenance activity?
Step 4: IMPLEMENT Corrective Action
Once the root cause of the out-of-control event has been identified, specific corrective action to address the problem can be performed. For example, if the root cause of the out-of-control event was degradation of the reagent stored onboard the instrument, a fresh container of reagent can be placed on the instrument. If the root cause of the out-of-control event was bacterial contamination of the instrument, then a decontamination procedure can be performed. Patient testing may resume if QC meets acceptance criteria after the corrective action is implemented. If QC does not meet acceptance criteria, then the root cause has not been determined and troubleshooting should continue.
It is not uncommon that laboratorians consider that the response to a QC out-of-range event ends with corrective action. They might think “Great! The problem is corrected and now patient sample testing can resume and we are finished addressing the out-of-control event.” However, there are still a number of steps remaining to recover from the out-of-control event.
Step 5: EVALUATE Impact on Previously Reported Patient Results
After corrective action, the laboratory should evaluate the impact of the error on previously reported patient results. If the root cause of the problem was determined to be degradation of the QC material, for example, then patient sample results would not have been impacted by the error since there was no problem with the measurement system. Most measurement system issues have the potential to impact patient sample results. If QC is analyzed every 12 hours, and an out-of-control event is detected, the actual measurement system problem could have happened at any point since the last acceptable QC result was obtained, and the laboratory could have already reported erroneous results into the medical record. Therefore, previously analyzed patient samples need to be reanalyzed and repeat results evaluated to determine if the magnitude of error could have caused a clinical impact, such as inappropriate patient diagnosis or therapy based on the erroneous results.
Once testing on the patient samples has been repeated, the repeat result (obtained after corrective action) should be compared with the original reported result to determine if the magnitude of the difference between results is large enough to adversely impact patient care. Acceptability criteria for the difference for patient sample repeats is established by the Laboratory Director and can be based on the allowable total error (TEa). The TEa is defined as the overall limit for acceptable imprecision and bias for the result of a single measurement, and is used to establish the maximum error that can be tolerated without adversely affecting medical decision-making. According to the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) conference2 recommendations there are three different models to determine the TEa for an assay. TEa can be based on clinical outcome studies, biological variation estimates3 (see: EFLM Biological Variation Database: https://biologicalvariation.eu/) or the state-of-the art assay performance. Other regulatory performance specifications can also be used, for example the Clinical Laboratory Improvement Amendments (CLIA) acceptable test performance criteria4. If the difference between results falls outside the TEa limits, then a clinically significant impact of the error is likely, and corrected reports need to be issued.
Step 6: Take Steps to MITIGATE PATIENT HARM
The next step of the recovery process is to quickly take steps to mitigate patient harm. Because patients may have received inappropriate clinical actions based on erroneous results, corrected reports should be immediately issued if the magnitude of error for the original reported results exceeds the TEa. Providers should be notified that a corrected report has been issued.
Some approaches to quickly mitigate patient harm include developing data entry templates to automatically identify patient results that require corrected reports and to call in extra staff to assist with patient sample repeat analyses and provider phone calls.
Step 7: IMPLEMENT PREVENTATIVE ACTION to Avoid Recurrence of the Out-of-Control Event
The last step of the recovery process is to implement preventative action to avoid recurrence of the event. Preventative action is distinct from corrective action. For example, if the out-of-control event was due to instability of an onboard reagent, the corrective action would be to replace the reagent with a fresh reagent container. The preventative action would be to change the onboard expiration of the reagent so that replacement of the reagent will occur more frequently, to prevent the problem from recurring. Procedure changes and staff training are frequently needed to ensure that preventative actions are implemented.
Other examples of preventative actions include:
- Increase QC analysis frequency to detect errors in a more timely manner and decrease the risk of reporting erroneous patient results, especially for unstable assays
- Adjust QC rule evaluation stringency to detect smaller deviations from the QC target mean
- Implement additional tools to evaluate assay performance such as inter-laboratory QC comparison programs, proficiency testing programs and patient-based real-time QC, e.g., moving patient averages
All steps of the response to the out-of-control event should be documented so the laboratory can refer to the root cause of the error, and corrective and preventative actions performed, in the event that a similar problem should happen again.
Summing up
No matter the cause, a systematic approach should be implemented by the laboratory to respond to an out-of-control event. The out-of-control event must first be detected by QC evaluation rules designed to identify when the measurement system deviates from normal performance. After detection of the event, reporting of patient results must be immediately stopped to avoid further reporting of potentially erroneous results that could adversely impact patient care. An investigation should be implemented to determine the root cause of the error, and corrective action should be performed.
However, the process does not end there. The impact of the error on patient results should be evaluated and measures should be taken to reduce potential patient harm. Finally, preventative action should be implemented to avoid recurrence of the error, and the entire process should be documented to enable continuous evaluation and improvement of the laboratory’s QC program.
About the Authors
Lorin M. Bachmann, PhD, DABCC, is professor of pathology and co-director of clinical chemistry at Virginia Commonwealth University, Richmond, Va.
Nico Vandepoele is scientific and professional affairs manager at Bio-Rad Laboratories, Irvine, Calif.
Featured Image: A graphical representation of the steps labs should take to recover from an out-of-control event. Illustration: Bio-Rad Laboratories
References
- CLSI. Statistical Quality Control for Quantitative Measurement Procedures: Principles and Definitions. 4th Edition. CLSI guideline C24. Wayne, PA: Clinical and Laboratory Standards Institute; 2016.
- Sandberg S, Fraser CG, Horvath AR, Jansen R, Jones G, Oosterhuis W, Hyltoft Pertersen P, Schimmel H, Sikaris K, Panteghini M. Defining analytical performance specifications: Consensus Statement from the 1st Strategic Conference of the European Federation of Clinical Chemistry and Laboratory Medicine. Clin Chem Lab Med 2015; 53(6): 833–835.
- Aarsand AK, et al. The EFLM biological variation database. https://biologicalvariation.eu/ (Accessed 9/01/21).
- CLIA. Requirement for analytical quality. Fed Reg 1992;57(40):7002–186.



{kind=link}